
Болезнь Марфана
Болезнь Марфана — это врожденная патология, характеризующаяся аномалиями развития соединительной ткани с преимущественным поражением сердца, сосудов, опорно-двигательного аппарата и органа зрения. Значительная доля нарушений связана с дисфункцией ферментных систем, регулирующих синтез структурных белков.
Впервые синдром Марфана был описан в 1876 году французским педиатром Вильямсом. Заболевание приводит к тяжелым расстройствам, инвалидности, а при отсутствии лечения — к быстрой смерти.
Классификация
Болезнь Марфана классифицируют в зависимости от генетической характеристики (семейная форма и первичная мутация), течения (стабильного или прогрессирующего), формы и клинической картины.
Заболевание может протекать в стертой или выраженной формах. Стертая форма характеризуется незначительно выраженными изменениями в одной или двух системах. При выраженных изменениях нарушения разной степени отмечаются в нескольких системах.
Клинические варианты заболевания:
- болезнь Марфана — наличие 3 основных признаков патологического процесса, семейная форма;
- синдром Марфана — наличие стертых форм с положительными диагностическими тестами;
- марфаноподобный синдром.
Причины синдрома Марфана
Синдром Марфана — наследственная болезнь соединительной ткани с аутосомно-доминантным типом наследования.
В основе заболевания лежит нарушение синтеза одного из белков соединительной ткани — фибриллина. Именно он придает соединительной ткани такие свойства как эластичность и сократимость.
Заболевание вызывает мутантный ген FBN1, кодирующий синтез гликопротеина белка фибриллина. Вследствие дефицита фибриллина или его аномального строения соединительная ткань обладает повышенной растяжимостью, и теряет способность выдерживать нормальные (физиологические) нагрузки. Приблизительно в 75 % случаев заболевание передается по наследству, а в 25 % вызывается спорадическими мутациями.
Патологические изменения в одном и том же участке (локусе) хромосомы могут вызывать различные клинические проявления — от стертой формы с поражением одной из систем организма до развернутых нарушений. Иногда клиническое разнообразие болезни Марфана обусловлено мутациями, локализованными в генах FBN2 или FBN3.
Клинические проявления
Соединительная ткань находится во многих органах, поэтому симптоматика болезни Марфана многосистемная и разнообразная.
Симптоматика заболевания проявляется поражением нескольких жизненно важных органов и целых систем: опорно-двигательного аппарата, сердечно-сосудистой и дыхательной систем, органов зрения, центральной нервной системы.
Признаки синдрома Марфана
В специфическую клиническую картину синдрома относят:
- астенический тип телосложения;
- высокий рост при дефиците массы тела;
- длинные и узкие пальцы (долихостеномелия);
- удлиненная и узкая голова (долихоцефалия);
- сколиоз;
- плоскостопие;
- деформированная грудная клетка – впалая (грудь “сапожника”) или выпуклая (“куриная” грудь);
- “птичье лицо” (большой нос и маловыраженный подбородок);
- сильное выступание верхней челюсти вперед, отсутствие контакта передних зубов обеих челюстей при смыкании (прогнатия).
Болезнь Марфана характеризуется мышечными болями, пониженным тонусом мышечной ткани, гипотонией кишечника. У больных часто отмечаются приступы сильной головной боли (особенно после эмоциональных потрясений и физических нагрузок).
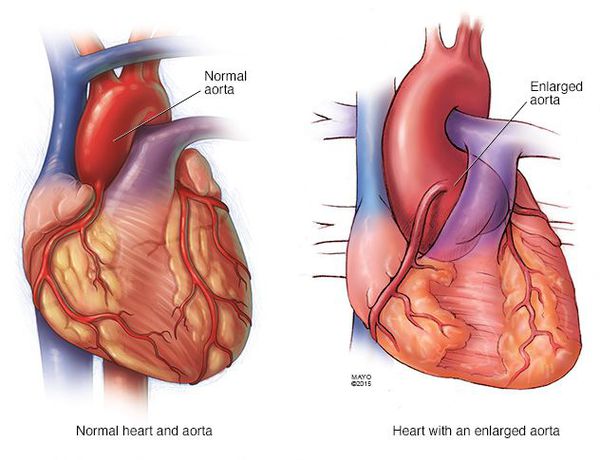
Тяжесть состояния при синдроме Марфана и прогноз зависят в основном от степени поражения сердца и сосудов, которые теряют способность выдерживать нормальные гемодинамические нагрузки. Уже на первом-втором году жизни ребенка регистрируются сердечно-сосудистые нарушения. Чаще всего наблюдается недостаточность митрального клапана. У больных постепенно увеличивается диаметр аорты, достигая критических размеров после 15 лет.
Поражения органов могут быть врожденными. К таким патологиям относят: недоразвитие легкого, врожденную буллезную эмфизему, поликистоз легких, бронхоэктатическую болезнь.
Коэффициент интеллектуального развития (IQ) у большинства детей обычно соответствует норме — 85-115 единиц. У некоторых лиц IQ превышает верхнюю границу нормы в 115 ед.
Отмечается своеобразие некоторых психических процессов и личностных особенностей (завышенная самооценка, раздражительность, плаксивость).
Дети с синдромом Марфана плохо переносят физические нагрузки, у них отмечаются нарушения показателей физического развития.
Осложнения
Патологические изменения соединительной ткани различных органов и систем при болезни Марфана ведет к развитию тяжелых заболеваний с летальным исходом.
Осложнения и последствия синдрома Марфана:
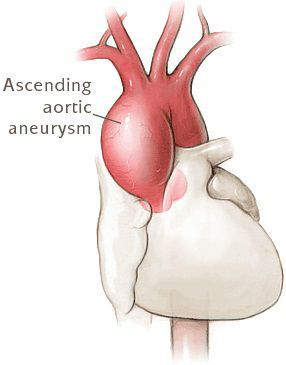
- Аневризма аорты и ее расслоение
- Сердечная недостаточная
- Дыхательная недостаточность
- Инфекционный эндокардит
- Пневмоторакс
- Эмфизема легких
- Кистозная болезнь легких
- Инфаркт легкого
- Вывих и подвывих хрусталика глаза
- Катаракта
- Косоглазие
- Близорукость
- Эктопия хрусталиков
- Мезодермальная дистрофия радужки
- Отслоение сетчатки глаза
Разрыв аневризмы аорты и сердечная недостаточность — основная причина гибели детей и взрослых с синдромом Марфана. Продолжительность жизни больных без лечения составляет 32 ± 16 лет. Раннее начало лечения позволяет значительно продлить жизнь больных и улучшить качество их жизни. При полноценном лечении продолжительность жизни увеличивается до 60 лет и более.
Диагностика
Диагностика заболевания основана на анализе родословных, генетических тестах, поиска лица, с которого началось заболевание (пробанда), наличия симптомов синдрома Марфана.
При постановке диагноза у ребенка педиатр изучает его физическое и нервно-психическое развитие, состояние отдельных органов и систем.
Оценка состояния организма проводится с помощью ряда фенотипических диагностических тестов, теста на арахнодактилию, определения индекса Дюранта-Лайнера, индекса телосложения Варги.
Для постановки диагноза необходимо наличие одного из пяти основных симптомов и двух из дополнительных.
Основные симптомы синдрома Марфана:
- вывих хрусталика;
- аневризма аорты;
- деформация грудной клетки;
- кифосколиоз;
- арахнодактилия (“паучьи пальцы” – аномально длинные и узкие пальцы по сравнению с ладонью).
К дополнительным диагностическим признакам относят: пролапс митрального клапана, миопия, умеренная гиперподвижность суставов, высокий рост, стрии (растяжки на коже), пневмоторакс.
Методы диагностики синдрома Марфана:
Диагностика синдрома Марфана включает консультации врача-ортопеда, кардиолога, осмотр офтальмолога.
Лечение
Лечение синдрома Марфана основано на комплексной терапии, включающей назначение лекарственных препаратов. При заболевании назначают средства, влияющие на сердечно-сосудистую систему, стимуляторы ЦНС, энерготропные препараты и антиоксиданты.
Немедикаментозные методы лечения синдрома Марфана
В такое лечение обычно входит:
- магнитотерапия на суставы;
- электросон; ;
- санаторно-курортное лечение.
Хирургические методы лечения
В хирургическое лечение входит:
- восстановление аорты и клапанов;
- тонзиллэктомия;
- аденотомия;
- экстракция хрусталика;
- эндопротезирование тазобедренного сустава;
- пластика грудной клетки;
- коррекция позвоночника.
Контроль излеченности
После комплексной терапии почти у 80 % детей с синдромом Марфана отмечается значительное улучшение. Об эффективности терапевтических мероприятий судят по определенным признакам. Среди них:
- повышение тонуса и объема мышц;
- стабилизация диаметра аорты;
- увеличение объема произвольной памяти;
- улучшение моторики;
- нормализация показателей внешнего дыхания;
- улучшение способности к концентрации.
Профилактика синдрома Марфана
Больным с синдромом Марфана, планирующим завести ребенка, показано генетическое консультирование с определением степени вероятности риска развития у детей данной патологии. Обязательно проводится пренатальная диагностика.
Советы и рекомендации
При наблюдении за больными с синдромом Марфана важно соблюдать определенные требования к режиму дня, отдыха и реабилитации.
Больным с синдромом Марфана нужно придерживаться таких правил:
- занятия физкультурой разрешены только в специальной группе (группы ЛФК и спецгруппы);
- запрещены занятия в спортивных секциях, участие в соревнованиях, походы на длительные дистанции по пересеченной и горной местности, поднятие тяжестей (более 3 кг);
- для постоянного проживания противопоказан жаркий климат.
Беременным с болезнью Марфана показана эхокардиография раз в 2 месяца. Если диаметр аорты достиг 45 мм и больше, рассматривается вопрос о возможности сохранения беременности.
Родоразрешение у женщин с синдромом Марфана осуществляют с помощью кесарева сечения в специализированных родильных домах для рожениц с патологией сердечно-сосудистой системы.
Статья носит информационно-ознакомительный характер. Пожалуйста, помните: самолечение может вредить вашему здоровью.

Заведующая педиатрическим отделением на Оболони, врач-педиатр высшей категории

Врач-педиатр, детский аллерголог высшей категории
Какой врач лечит синдром Марфана?
Педиатры, врачи-кардиологи, детские ортопеды, генетики клиники МЕДИКОМ предлагают новейшие эффективные методики в лечении синдрома Марфана в Киеве (районы Оболонь и Печерск). Записаться на консультацию и получить всю интересующую информацию вы можете по одному из телефонов, указанных на сайте клинике.
Синдром Марфана – симптомы и лечение
Что такое синдром Марфана? Причины возникновения, диагностику и методы лечения разберем в статье доктора Боровиковой Ольги Игоревны, генетика со стажем в 7 лет.
Над статьей доктора Боровиковой Ольги Игоревны работали литературный редактор Маргарита Тихонова , научный редактор Сергей Федосов

Определение болезни. Причины заболевания
Синдром Марфана (Marfan; СМ) — генетически обусловленное заболевание, при котором происходит системное поражение соединительной ткани. [1]
Причины синдрома Марфана
Этиологией заболевания является мутация в гене FBN1 (фибриллина 1), расположенном в коротком плече пятнадцатой хромосомы в локусе 21.1. [2]
Тип наследования синдрома — аутосомно-доминантный. Для болезни характерна высокая пенетрантность (частота появления гена) и различная экспрессивность. [5]
Соотношение представителей мужского пола и женского одинаковое.
При обнаружении схожих симптомов проконсультируйтесь у врача. Не занимайтесь самолечением – это опасно для вашего здоровья!
Симптомы синдрома Марфана
Наблюдается постоянно прогрессирующее развитие заболевания. У новорожденных детей выявляются удлинённые тонкие пальцы на верхних и нижних конечностях и удлинённые тонкие конечности (долихостеномелия). [1] У таких пациентов, помимо долихостеномелии, отмечается:
- повышенное физическое развитие;
- недостаток веса;
- удлинённый череп;
- вытянутое лицо;
- арахнодактилия (аномально удлинённые узкие пальцы);
- слабость и недоразвитие мышечной системы и жировой клетчатки;
- неловкие движения. [3]
Кожа имеет повышенную растяжимость, разболтанные суставы. У большинства больных наблюдается высокое аркообразное нёбо, изменения формы грудной клетки (воронкообразная, килевидная) и искривления позвоночника (сколиоз в 60%, кифоз (изгиб позвоночника с образованием горба), ювенильный остеохондроз), уплощение свода стопы, аускультативные признаки порока сердца (шумы). [4] Длина третьего пальца руки — 10 см и больше (скрининговый тест у детей 7-18 лет): возрастает соотношение размаха верхних конечностей к длине тела.
Офтальмологические симптомы (близорукость, подвывих хрусталика в 75% случаев, его округлость или гипоплазия, отслойка сетчатки) и астенические признаки (усталость, вялость) обращают на себя внимание со второго года жизни, изменения формы грудной клетки появляются в возрасте старше четырёх лет, патология сердца и сосудов выявляется в дошкольном возрасте. [1]
Почти у всех больных выявляются пороки сердца и аорты. Часты бедренные и паховые грыжи, поражение клапанов в венах, их варикозное расширение, геморрагический синдром, рецидивирующие вывихи, поражение лёгочной системы (самопроизвольный пневмоторакс, эмфизематозное расширение лёгких), опущение почек. [2]
В четверти случаев зарегистрировано снижение интеллекта, у половины пациентов выявляются нарушения эмоционально-волевой сферы. Часто появляются депрессивные состояния, нейроциркуляторная дистония. [3]
По данным многих исследований, абсолютное большинство больных с синдромом Марфана отмечают ухудшение эмоционального фона, утрату чувства радости и увлечённости профессиональной деятельностью, частую смену настроения, повышенную возбудимость, чувство тревоги. Результатом этого является снижение социальной активности, ухудшение качества жизни и значительное уменьшение социальной адаптации. [3]
У таких пациентов часто наблюдается трахеобронхиальная дискинезия (нарушение дыхательной системы) за счёт слабости соединительнотканного каркаса бронхов. Это проявляется рецидивирующими воспалительными заболеваниями бронхолегочной системы, обструктивными нарушениями, бронхиальной астмой, эмфиземой лёгких (повышенное содержание воздуха в лёгочной ткани). [4] Встречаются осложнения, которые проявляются скоплением воздуха в грудной клетке, сопровождающиеся сдавлением лёгких и средостения (срединной области грудной клетки), подкожной эмфиземой. Наблюдается неадекватный ответ на бронхолитики. Обструктивные явления (непроходимость) затрагивают преимущественно верхние отделы респираторного тракта. [3]
Описаны характерные изменения на электрокардиограмме, включающие синдром раннего возбуждения желудочков, преждевременные желудочковые комплексы, нестабильность конечной части желудочкового комплекса в задненижних отведениях. [3]
Патология ритма чаще всего проявляются блокадой правой ножки пучка Гиса или смешанной экстрасистолией. [6]
У больных синдромом Марфана с патологией ритма сердечной деятельности и проводимости синдром вегетативной дисфункции чаще протекает по ваготоническому типу, в виде пресинкопальных, обморочных и астеновегетативных состояний, болезненных ощущений в области сердца, цефалгии напряжения (головной боли) и зачастую сочетается с психопатологическими расстройствами. [4]
Органы пищеварения также задействованы в патологическом процессе, что проявляется дискинезией (нарушением моторики) билиарного тракта со снижением моторики гладкомышечной мускулатуры, недостаточностью кардии, грыжевыми выпячиваниями пищеводного отверстия диафрагмы, аномалиями желчевыводящих протоков, долихосигмой (увеличением сигмовидной кишки), хроническим гастродуоденитом (воспалением слизистой желудка и двенадцатиперстной кишки), дисбиозом (нарушением нормальной микрофлоры) кишечника, изменениями поджелудочной железы. [3]
У пациентов с синдромом Марфана чаще, чем у здоровых людей, встречаются приобретённые аномалии почек: повышенная подвижность почек, нефроптоз (опущение почки), пиелоэктазии (аномальное расширение лоханок), повышена частота удвоения почек.
Патогенез синдрома Марфана
Более половины веса человека представлено соединительной тканью, из неё состоит наша главная опора — скелет, внешние покровы — кожа. Сосуды, кровь и лимфа тоже состоят из соединительной ткани.
К клеткам соединительной ткани относятся фибробласты и их разновидности (остеобласты, хондроциты, одонтобласты, кератобласты), макрофаги (гистиоциты) и тучные клетки (лаброциты). [7]
Мезенхима — проводник конституциональных, генетических и эпигенетических составляющих жизни человека. Патология соединительной ткани детерминирует определенное патологическое действие на весь организм в целом, на его физиологию и его конституциональные особенности. [3]
При болезни Марфана происходит замена нуклеотидов в гене, содержащем информацию о структуре пептида фибриллина-1. Этот белок относится к гликопротеидам, принимает участие в микрофибриллярном комплексе, он обеспечивает основу эластических фибрилл соединительной ткани.
Межклеточный матрикс позволяет соединительной ткани поддерживать постоянную структуру, в нём находится огромное количество факторов роста, которые обеспечивают постоянное обновление клеток.
В крупных сосудах, связочном аппарате содержится большое количество эластиновых фибрилл, поражение которых и даёт основные клинические проявления синдрома Марфана.
При синдроме Марфана значительно поражается трансформирующий фактор роста бета (TGF-β), нарушается связывание его неактивной формы, что приводит к повышению биоактивности данного фактора, с чем связано появление многих проявлений болезни. [4]
Патология фибриллина приводит к патологии формирования волокон, что вызывает утерю прочности и эластичности кожи и других соединительнотканных структур.
Изменение структуры коллагеновых волокон приводит к нарушению первичного звена гемостаза у пациентов с синдромом Марфана. [6]
Имеются данные о дефектах мембранных и цитоплазматических механизмов проведения сигнала непосредственно в самом тромбоците, приводящих к нарушениям агрегации (объединения). Показано наличие самостоятельного мембранного дефекта тромбоцитов, протекающего с нарушением реакций высвобождения и транспорта внутриклеточного кальция. [6]
Эластические фибриллы имеют вполне определенные механизмы участия в системе гемостаза. В сосудах с низкой скоростью сдвига происходит адгезия («прилипание») тромбоцитов к эластину через фибронектин. [7] Регистрируется снижение его уровня в крови у людей с синдромом Марфана. Фибронектин, в свою очередь, образуется в клетках эндотелия и участвует в последующих репаративных процессах, создавая основу для производства других компонентов соединительной ткани — фибробластов. [4] Таким образом, совершенно неоспоримо участие сосудистой стенки в реакциях свертываемости крови, и неизбежен вывод о возможных патологиях протекания нормальных гемостатических процессов при изменении состояния её структурных компонентов и процессов сосудистой регуляции.
Отмечена роль гормонального дисбаланса в развитии и усугублении дефектов соединительнотканных структур. [3]
Тромботические проявления детерминированы нарушением реологии (вязкости) крови в патологически извитых сосудах брахиоцефальной зоны. [3]
Поражение желудочно-кишечного тракта детерминировано тем, что эта система богата коллагеном. Наблюдаются дискинезия билиарного тракта по гипомоторному типу, грыжи пищеводного отверстия диафрагмы, аномалии желчных путей, долихосигма, хронический гастродуоденит со стёртой клинической картиной, склонностью к торпидному течению. [3]
Классификация и стадии развития синдрома Марфана
Код синдрома Марфана по Международной классификации болезней (МКБ-10): Q87.4.
- стёртая (поражено не более двух систем, изменения выражены незначительно);
- выраженная (незначительные изменения в трёх системах либо значительное поражение одной и более систем).
Выделяют различные типы по степени тяжести:
Частота тяжёлых форм — 1 к 25000-50000 (при общей частоте диагностированных случаев 1 к 10000-15000).
По характеру течения:
- прогрессирующая форма;
- стабильная форма.
Чаще всего первые признаки синдрома Марфана проявляются еще в детском периоде, с возрастом происходит прогрессирование симптомов, усиление клинических проявлений.
Осложнения синдрома Марфана
К самым частым осложнениям синдрома Марфана относятся:
- Снижение зрения, вплоть до слепоты, обусловленное слабостью цинновой связки (ресничного пояска) и подвывихом, вывихом хрусталика. [7]
- Сердечная недостаточность по застойному типу, обусловленная нарушением сократимости сердечной мышцы, недостаточностью митрального клапана. [6]
- Разрывы крупных сосудов, связанные с дилатацией (расширением), истончением стенки сосудов. Чаще всего происходит поражение аорты (в основном из-за изменения гемодинамики при беременности). [7]
- Расслаивающая аневризма аорты, приводящая к смерти больных.
Диагностика синдрома Марфана
Диагностика Синдрома Марфана основывается на клинических данных, выявлении изменений в гене FBN1. [5]
Часто при сборе генеалогического анамнеза выявляются родственные случаи со скрытым течением заболевания. [1]
Способы обнаружения арахнодактилии: [3]
- Симптом Steinberg (признак первого пальца). Первый палец виден из-под hypothenar при напряжённом кулаке.
- Симптом Walker-Murdoch (признак запястья). При обхватывании кистью в области лучезапястного сочленения контралатеральной верхней конечности первый палец заходит за пятый.
- Определение пястного индекса. Определяется при помощи рентгенографии. Средняя длина пясти, делённая на усреднённую ширину отрезка от второй до четвертой пястной кости. При нормальном соотношении этот показатель соответствует 5,4-7,9, в то время, как при синдроме Марфана — больше 8,4.
В 2010 году группа специалистов систематизировала международные Гентские критерии для верификации синдрома Марфана. Верификация зависит от данных генеалогического анамнеза. [3]
При отсутствии генеалогического анамнеза:
- увеличение диаметра аорты >, = 2 ϭ + эктопия хрусталика = СМ;
- увеличение диаметра аорты >, = 2 ϭ + выявленные изменения в гене FBN1 = CM;
- увеличение диаметра аорты >, = 2 ϭ + >, = 7 системных признаков = СМ;
- эктопия хрусталика + наличие изменений в гене FBN1 + дилатация аорты = СМ;
При наличии генеалогического анамнеза:
- Эктопия хрусталика + случай СМ в семье = СМ;
- >, = 7 системных проявлений + случай СМ в семье = СМ;
- увеличение диаметра аорты >, = 2 ϭ + случай СМ в семье = СМ.
В пятнадцати процентах появление ребёнка с синдромом Марфана спорадическое (случайное), у родителей могут быть слабые проявления. У родственников пациентов встречаются заболевания желудочно-кишечного тракта, поражения позвоночника, заболевания глаз. [3]
При малейшем подозрении на синдром Марфана необходима консультация офтальмолога. В анализе мочи таких пациентов отмечается повышение уровня оксипролина, гликозаминогликанов, но эти показатели низкоспецифичны, могут быть при различных дисплазиях соединительной ткани. Выделение оксипролина является показателем тяжести заболевания. Наблюдается нарушение свертываемости крови на тромбоцитарном уровне. [3]
Оценка системных признаков вовлечённости соединительной ткани
Синдром Марфана
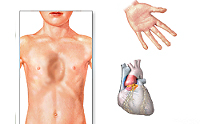
Синдром Марфана – дифференцированная форма врожденной соединительнотканной недостаточности, характеризующаяся разнообразными проявлениями скелетной, сердечно-сосудистой и глазной патологии. У больных с синдромом Марфана отмечаются гигантизм, долихостеномелия и арахнодактилия, аневризмы аорты, миопия, эктопия хрусталика, деформация грудины, кифосколиоз, плоскостопие, протрузия вертлужной впадины, эктазия твердой мозговой оболочки. Диагноз синдрома Марфана основан на семейном анамнезе, результатах функционального, офтальмологического, рентгенологического и генетического исследований. Лечение при синдроме Марфана включает консервативную и хирургическую коррекцию сердечно-сосудистых нарушений, поражений скелета и органа зрения.
МКБ-10
Общие сведения
Синдром Марфана – системное недоразвитие соединительной ткани в эмбриональном и постнатальном периодах, обусловленное структурными дефектами коллагена и сопровождающееся преимущественным поражением опорно-двигательного аппарата, глаз, сердечно-сосудистой системы. Синдром Марфана – одна из наиболее распространенных наследственных коллагенопатий синдромального характера. Частота встречаемости синдрома Марфана в популяции невысока: по данным различных авторов составляет 1 случай на 10000-20000 человек, без расовой и половой детерминированности.
Причины синдрома Марфана
Синдром Марфана относится к врожденным аномалиям, наследуемым по аутосомно-доминантному типу, с выраженным плейотропизмом, варьирующей экспрессивностью и высокой пенетрантностью. В основе синдрома Марфана лежат мутации в гене FBN1, отвечающем за синтез фибриллина – важнейшего структурного белка межклеточного матрикса, придающего эластичность и сократимость соединительной ткани. Аномалия и дефицит фибриллина при синдроме Марфана приводят к нарушению формирования волокнистых структур, потере прочности и упругости соединительной ткани, невозможности выдерживать физиологические нагрузки. Гистологическим изменениям в большей степени подвержены стенки сосудов эластического типа и связочный аппарат (в первую очередь, аорта и цинновая связка глаза, содержащие наибольшее количество фибриллина).
Широкий фенотипический спектр синдрома Марфана (от легких форм, трудно отличимых от нормы до тяжелых, быстропрогрессирующих) объясняется разнообразием мутаций в гене FBN1 (более 1000 видов), а также присутствием мутаций в других генах (например, в гене трансформирующего фактора роста – TGFBR-2). При генетическом исследовании в 75% случаев синдрома Марфана выявляется семейный тип наследования, в остальных – первичная мутация. Риск рождения ребенка с синдромом Марфана возрастает с увеличением возраста отца (особенно после 35 лет).
Классификация синдрома Марфана
В зависимости от количества пораженных систем выделяют несколько форм синдрома Марфана:
- стертую – со слабо выраженными изменениями в 1-2-х системах
- выраженную – со слабо выраженными изменениями в 3-х системах; выраженными изменениями хотя бы в 1-ой системе; выраженными изменениями в 2-3-х и более системах.
Степень тяжести изменений при синдроме Марфана может быть легкой, средней и тяжелой. По характеру течения дифференцируют прогрессирующий и стабильный синдром Марфана.
Симптомы синдрома Марфана
Синдром Марфана характеризуется сочетанным поражением скелета, глаз, сердечно-сосудистой и нервной систем; многообразием проявлений, варьированием сроков появления первых признаков заболевания; хроническим прогредиентным течением.
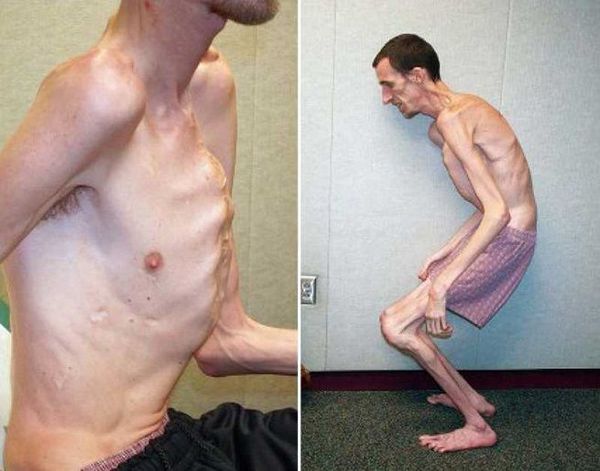
Больные синдромом Марфана, как правило, отличаются высоким ростом, относительно коротким туловищем с непропорционально длинными тонкими конечностями (долихостеномелией) и удлиненными паукообразными пальцами (арахнодактилией); астеническим телосложением со слаборазвитой подкожной клетчаткой и мышечной гипотонией; длинным и узким лицевым скелетом (долихоцефалией); наличием высокого аркообразного неба и нарушения прикуса (прогнатии). Средняя длина тела при рождении у мальчиков с синдромом Марфана составляет 53 см, окончательный рост – 191 см; у девочек – соответственно 52,5 см и 175 см.
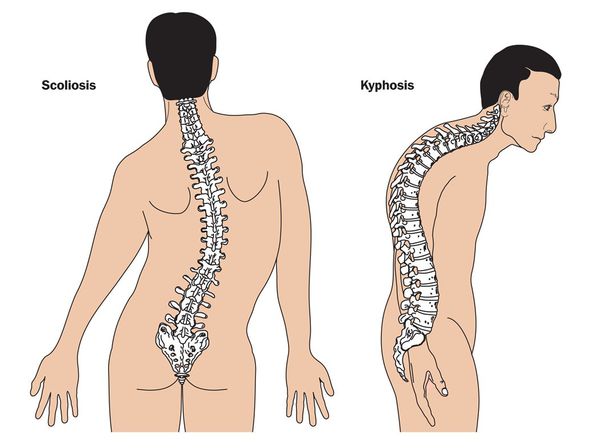
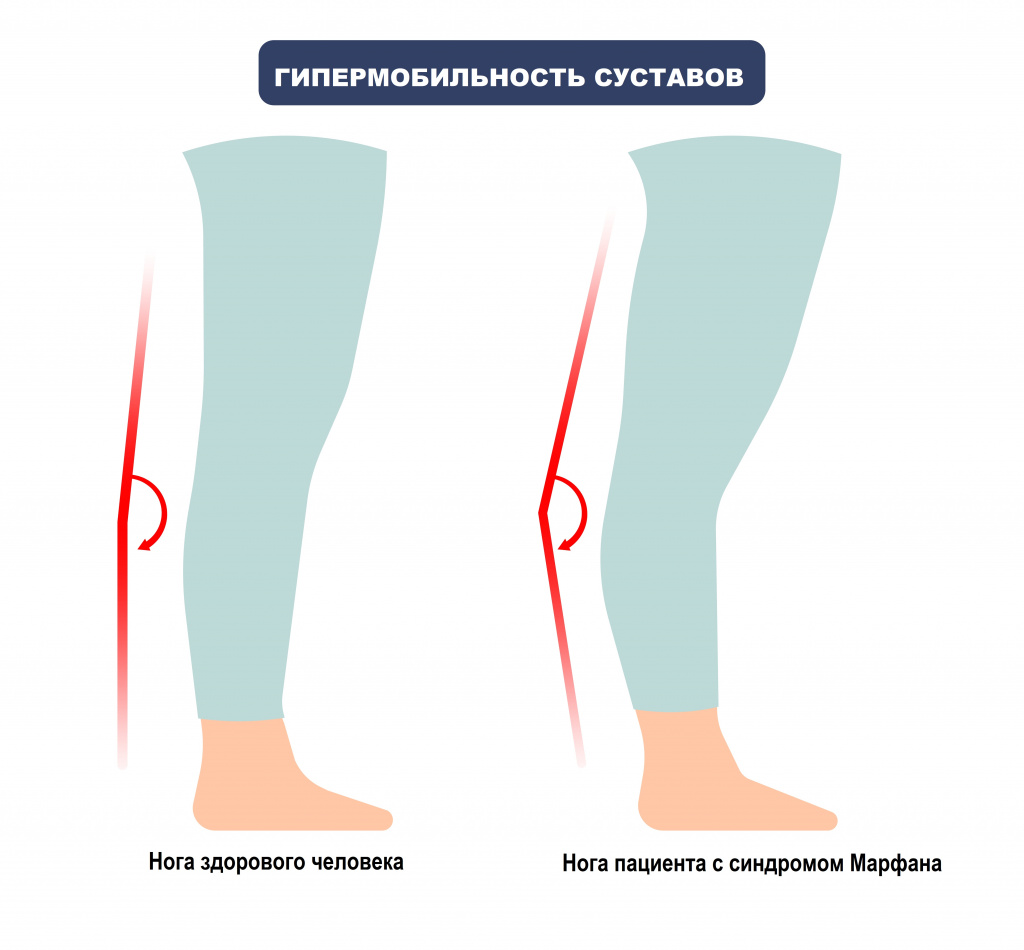
При синдроме Марфана отмечаются нарушение функции суставов (гипермобильность); деформация грудной клетки (воронкообразная или килевидная форма), деформация позвоночника (сколиоз, кифоз, кифосколиоз, подвывихи и вывихи шейного отдела, спондилолистез), а также плоскостопие и протрузия вертлужной впадины.
Сердечно-сосудистая патология, доминирующая в клинической картине синдрома Марфана и часто определяющая его исход, проявляется дефектами структуры стенок сосудов эластического типа, особенно аорты и крупных ветвей легочной артерии, пороками развития клапанного аппарата и перегородок сердца. Изменения аорты у больных синдромом Марфана характеризуются прогрессирующим расширением ее восходящей части и клапанного кольца (дилатацией, аннулоаортальной эктазией) и аневризмами; поражение митрального клапана – миксоматозной дегенерацией створок, патологическим удлинением и разрывом створочных хорд, обызвествлением клапанного кольца. У плода с синдромом Марфана возможно формирование врожденных пороков сердца – коарктации аорты, стеноза легочной артерии, ДМПП и ДМЖП. Органические и функциональные изменения сердца и сосудов у больных синдромом Марфана часто сопровождаются нарушением ритма (наджелудочковой и желудочковой тахикардией, фибрилляцией предсердий) и развитием инфекционного эндокардита.
Самая неблагоприятная неонатальная форма синдрома Марфана проявляется в классическом варианте уже при рождении, приводит к прогрессирующей сердечной недостаточности и летальному исходу на первом году жизни ребенка.
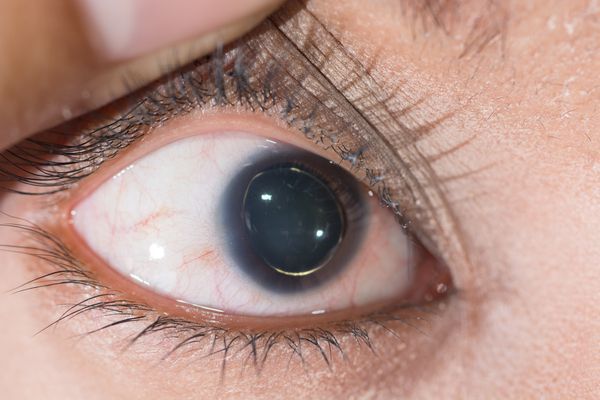
Для большинства случаев синдрома Марфана характерна патология органа зрения, включающая близорукость, вывих/подвывих (эктопию) хрусталика, уплощение и увеличение размера роговицы, гипоплазию радужной оболочки и цилиарной мышцы, косоглазие, изменение калибра сосудов сетчатки. Эктопия хрусталика при синдроме Марфана имеет двухсторонний характер, часто развивается в возрасте до 4-х лет и устойчиво прогрессирует, ухудшая зрительную функцию.
При синдроме Марфана наблюдается поражение других систем и органов: нервной (эктазия твердой мозговой оболочки, в т. ч. пояснично-крестцовое менингоцеле), бронхолегочной (спонтанный пневмоторакс, эмфизема легких, дыхательная недостаточность), кожи и мягких тканей (атрофические стрии), рецидивирующие паховые и бедренные грыжи, вывихи и разрывы связок, а также эктопия почек, опущение мочевого пузыря и матки, варикозное расширение вен и др.
Характерный для синдрома Марфана высокий выброс адреналина может способствовать постоянному нервному возбуждению, гиперактивности, а иногда развитию неординарных способностей и умственной одаренности.
Диагностика
Диагноз синдрома Марфана основывается на семейном анамнезе, наличии у больного типичных диагностических признаков по результатам физикального осмотра, ЭКГ и ЭхоКГ, офтальмологического и рентгенологического обследования, молекулярно-генетического анализа и лабораторных исследований.
За диагностические критерии синдрома Марфана берутся характерные изменения в различных системах и органах; главными (большими) из них считаются: дилатация корня/расслоение восходящей части аорты, эктопия хрусталика и эктазия твердой мозговой оболочки; килевидная/воронкообразная деформация грудной клетки, требующая хирургического лечения; отношение длины верхнего сегмента тела к нижнему 1,05; сколиоз (> 20˚) или спондилолистез; ограничение разгибания в локтевом суставе (
Также применяются фенотипические диагностические тесты, определяющие соотношение кисть/рост (при синдроме Марфана > 11%); длину среднего пальца (> 10 см); индекс телосложения Варги – (масса тела, г/(рост, см)x2 – возраст, годы/100, должно быть
ЭКГ при синдроме Марфана позволяет определить нарушение ритма сердца, выраженную гипертрофию миокарда; ЭхоКГ – обнаружить клапанную регургитацию, увеличение размеров левого желудочка, пролапс митрального клапана, разрывы хорд, дилатацию аорты. На рентгенографии грудной клетки можно увидеть расширение корня и дуги аорты, увеличение размеров сердца; на КТ и МРТ сердца и сосудов – выявить дилатацию и аневризмы аорты.
Аортография показана при подозрении на аневризму и расслоение аорты. Наличие эктопии хрусталика уточняют с помощью биомикроскопии и офтальмоскопии; протрузию вертлужной впадины устанавливают методом рентгенографии тазобедренных суставов; эктазию твердой мозговой оболочки – МРТ позвоночника.
При синдроме Марфана определяется возрастание (в 2 раза и более) почечной экскреции метаболитов соединительной ткани: глюкозоаминогликанов и их фракций. Метод прямого автоматического секвенирования ДНК позволяет провести генетическую идентификацию мутаций в гене FBN1.
Необходима дифференциальная диагностика с заболеваниями, внешне напоминающими синдром Марфана: гомоцистинурией, врожденной контрактурной арахнодактилией (синдромом Билса), наследственной артроофтальмопатией (синдромом Стиклера), MASS-синдромом, синдромами Элерса-Данлоса, Лойса-Дитца, Шпринцена–Голдберга, семейной эктопией хрусталика и др.
Лечение синдрома Марфана
Лечение и дальнейшее наблюдение пациентов с синдромом Марфана должно осуществляться группой специалистов: офтальмологом, кардиологом, кардиохирургом, ортопедом, генетиком, терапевтом.
Лечение больных с синдромом Марфана направлено на профилактику прогрессирования заболевания и развития осложнений, в первую очередь в сердечно-сосудистой системе. При диаметре аорты до 4 см назначаются β-адреноблокаторы, антагонисты кальция или ингибиторы АПФ. Хирургическое лечение проводится при недостаточности клапанов сердца, пролапсе митрального клапана, значительном расширении (>5 см) восходящей части и расслоении аорты. Реконструктивные операции на аорте при синдроме Марфана, имеют высокий процент послеоперационной 5-ти и 10-ти летней выживаемости. При необходимости выполняют протезирование митрального клапана. У беременных с синдромом Марфана и выраженной сердечно-сосудистой патологией проводят досрочное оперативное родоразрешение путем кесарева сечения. С целью профилактика инфекционного эндокардита и тромбозов после операционных вмешательств назначаются антибиотики и антикоагулянты.
При синдроме Марфана проводится коррекция зрения с помощью подбора очков и контактных линз, при необходимости – лазерное или хирургическое лечение катаракты, глаукомы, удаление смещенного хрусталика с имплантацией искусственного. При выраженных скелетных нарушениях может потребоваться хирургическая стабилизация позвоночника, торакопластика, эндопротезирование тазобедренных суставов. Применяются также патогенетическая коллагеннормализующая терапия, метаболическая и витаминотерапия.
Прогноз
Прогноз жизни больных с синдромом Марфана определяется, в первую очередь, степенью сердечно-сосудистых изменений, а также поражений скелета и глаз. Имеется высокий риск осложненного течения, снижения продолжительности жизни (90-95% не доживают до 40-50 лет) и внезапной смерти. Своевременная кардиохирургическая коррекция при синдроме Марфана позволяет значительно увеличить продолжительность (до 60-70 лет) и улучшить качество жизни больных.
Больные синдромом Марфана должны находиться под постоянным врачебным наблюдением и регулярно проходить диагностическое обследование. При синдроме Марфана показан низкий или средний уровень физической активности, исключающий занятия контактными видами спорта, спортивные соревнования, изометрические нагрузки, подводное плавание. Женщинам детородного возраста с синдромом Марфана необходимо пройти медико-генетическое консультирование.
Синдром Марфана: признаки, диагностика, лечение, причины, продолжительность жизни
Этиология и встречаемость синдрома Марфана. Синдром Марфана (MIM №154700) — панэтническое аутосомно-доминантное заболевание соединительной ткани, вызванное мутациями в гене фибриллина 1 (FBN1, MIM № 134797).
Синдром Марфана имеет встречаемость около 1 на 10 000. Приблизительно 25-35% пациентов имеют новую мутацию. Мутации, вызывающие синдром Марфана, разбросаны по гену, и каждая мутация обычно уникальна в семье.
Патогенез синдрома Марфана
Ген FBN1 кодирует фибриллин 1, внеклеточный матричный гликопротеид с широким распределением. Фибриллин 1 полимеризуется, формируя микрофибриллы как в эластичных, так и в неэластичных тканях, например стенке аорты, цилиарных поясках и коже.
Мутации влияют на синтез, процессинг, секрецию, полимеризацию или устойчивость фибриллина 1. Исследования накопления и экспрессии фибриллина 1 в культуре клеток предположили доминантный отрицательный патогенез, т.е. синтез мутантного фибриллина 1 тормозит образование нормальных микрофибрилл нормальным фибриллином 1 или стимулирует протеолиз несоответствующих внеклеточных микрофибрилл.
Последние исследования на мышиных моделях синдрома Марфана указывают, что половинного количества нормального фибриллина 1 недостаточно, чтобы проводить эффективную сборку микрофибрилл. Таким образом, патогенезу болезни также может содействовать гаплонедостаточность.
Кроме синдрома Марфана, мутации в гене FBN1 могут вызывать другие синдромы, включая неонатальный синдром Марфана, изолированные скелетные симптомы, аутосомно-доминантную эктопию хрусталиков и фенотип MASS (марфаноидные симптомы, включая пролапс митрального клапана или миопию, пограничное и непрогресирующее расширение аорты, и неспецифические изменения скелета и кожи).
В общем, фенотипы довольно схожи в пределах семьи, хотя тяжесть фенотипических проявлений может значительно изменяться. До настоящего времени точное соотношение между генотипом и фенотипом не определено. Внутрисемейная и межсемейная изменчивость позволяет предполагать, что в определении фенотипа важную роль играют окружающая среда и эпигенетические факторы.
Последние исследования на мышиных моделях показывают, что фибриллин 1 не просто структурный белок, и что синдром Марфана не просто результат структурной слабости тканей. Более того, микрофибриллы фибриллина 1 в норме связывают и уменьшают концентрацию и активность факторов роста суперсемейства TGFb.
Потеря фибриллина 1 увеличивает сигналы свободного TGFb значительно содействующие заболеванию, так как антагонисты TGFb устраняют легочные и клапанные изменения, наблюдаемые у мышей с недостаточностью фибриллина 1.
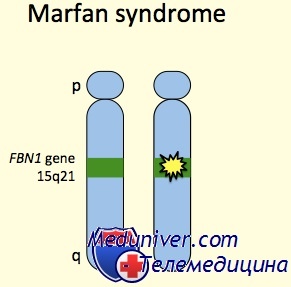
Фенотип и развитие синдрома Марфана
Синдром Марфана — мультисистемное заболевание со скелетными, глазными, сердечно-сосудистыми, легочными, кожными и другими аномалиями. Скелетные аномалии включают очень высокий рост (отношение размаха рук к росту >1,05; соотношение верхнего и нижнего сегментов
Аномалии глаз включают подвывих хрусталиков, уплощение роговицы, удлинение глазного яблока и гипоплазию радужки. Сердечнососудистые аномалии включают пролапс митрального клапана, аортальную регургитацию и расширение и расслаивающую аневризму восходящей аорты. Легочные аномалии включают спонтанный пневмоторакс и расширение концевых пузырьков. Аномалии кожи включают атрофические бороздки и рецидивирующие грыжи. Аномалии твердой мозговой оболочки включают выбухание оболочки в крестцово-поясничном отделе.
Большинство признаков синдрома Марфана появляются с возрастом. Скелетные аномалии типа аномалии грудины и сколиоза ухудшаются с ростом костей. Подвывих хрусталика часто присутствует уже в раннем детстве, но может развиваться и в юности.
С повышенной частотой при синдроме Марфана встречаются отслойка сетчатки, глаукома и катаракты. Сердечно-сосудистые осложнения обнаруживаются в любом возрасте и развиваются в течение всей жизни.
Основные причины преждевременной смерти пациентов с синдромом Марфана — сердечная недостаточность вследствие регургитации клапанов и аневризмы и разрыва аорты. Тем не менее в связи с улучшением хирургической и терапевтической помощи при аневризме аорты выживание улучшилось. С 1972 по 1993 г. ожидаемый возраст выживания для 50% пациентов поднялся с 49 до 74 лет для женщин и с 41 до 70 лет для мужчин.
Особенности фенотипических проявлений синдрома Марфана:
• Возраст начала: раннее детство
• Несоразмерно высокий рост
• Скелетные аномалии
• Эктопия (подвывих) хрусталика
• Пролапс митрального клапана
• Аневризма и разрыв аорты
• Спонтанный пневмоторакс
• Грыжа оболочки мозга в пояснично-крестцовом отделе
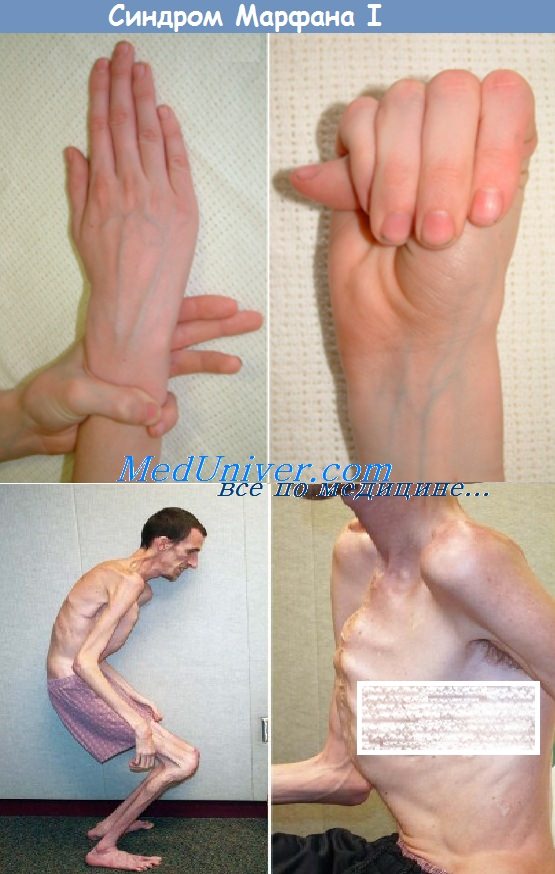
Лечение синдрома Марфана
Синдром Марфана — клинический диагноз, определяемый по наличию конкретных симптомов. Подтверждение синдрома Марфана идентификацией мутаций в гене FBN1 в настоящее время практически нецелесообразно, поскольку крайняя аллельная гетерогенность делает идентификацию причинно-обусловленной мутации в каждой семье крайне трудозатратной, а также из-за недостаточно надежной корреляции между генотипом и фенотипом. Анализ мутаций имеет недостаточную чувствительность или специфичность для синдрома Марфана, что ограничивает его клиническую пользу.
Для синдрома Марфана недоступно эффективное лечение; следовательно, помощь сфокусирована на профилактике осложнений и симптоматическом лечении. Оказание офтальмологической помощи включает регулярные осмотры, коррекцию миопии и, часто, замену хрусталика. Ортопедическая помощь заключается в укрепляющем лечении или хирургической коррекции сколиоза. Помощь при аномалии грудины в основном косметическая.
Физиотерапия может компенсировать нестабильность суставов. Сердечно-сосудистые проблемы решаются комбинацией терапевтических и хирургических мероприятий. Терапевтические усилия направлены на предохранение или замедление развития расширения корня аорты за счет уменьшения кардиологических показателей, снижения артериального давления и усилия выброса желудочков с помощью бета-адреноблокаторов, ограничение участия в контактных видах спорта, соревновательных видах спорта и в изометрических упражнениях.
Профилактическая замена корня аорты показана, когда расширение аорты или аортальная регургитация становится достаточно тяжелой. Большинству пациентов в настоящее время проводят надклапанную замену корня аорты, не требующую постоянного приема противосвертывающих препаратов.
Гемодинамические изменения, связанные с беременностью, могут приводить к прогрессирующему расширению или расслоению аорты. Полагают, что расслоение аорты вторично к гормональным изменениям, увеличению объема крови и сердечного выброса, связанных с беременностью и родами. Современные исследования считают, что риск беременности слишком велик, если ширина корня аорты превышает 4 см. Женщины могут выбрать проведение надклапанной замены аорты перед беременностью.
Риски наследования синдрома Марфана
Пациенты с синдромом Марфана имеют 50% риск иметь ребенка, больного синдромом Марфана. В семьях, передающих синдром Марфана, членов семьи, находящихся в группе риска, можно выявлять, либо обнаруживая мутацию (в тех редких случаях, когда она известна), либо анализом сцепления, если маркеры, тесно сцепленные с локусом FBN1, имеют очевидную связь с болезнью в семье пробанда. Пренатальная диагностика доступна только для семей, в которых возможны исследования сцепления или известна мутация в гене FBN1.
Пример синдрома Марфана. Д.Л., здоровый 16-летний ученик средней школы, звезда баскетбола, направлен в генетическую клинику для обследования по поводу синдрома Марфана. Телосложение Д.Л. похоже на телосложение его отца. Отец Д.Л., высокий субтильный человек, умер во время утренней пробежки; у других членов семьи случаев скелетных аномалий, внезапной смерти, снижения зрения или врожденных аномалий не было.
При медицинском осмотре выявлены астеническое телосложение, высокое дугообразное нёбо, небольшая деформация грудины по типу «куриной» груди, арахнодактилия, соотношение размах рук/рост 1,1, диастолический шум и стрии на плечах и бедрах. Эхокардиография выявила расширение корня аорты с аортальной регургитацией. Офтальмологическое обследование показало двусторонний иридодонез и легкое смещение хрусталиков кверху. На основе медицинского осмотра и результатов обследования генетик объяснил пациенту и его матери, что у него синдром Марфана.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Синдром Марфана

Синдром Марфана — наследственное заболевание, которое проявляется системным поражением соединительной ткани в организме человека. В результате болезни происходят нарушения строения скелета и кожи, работы глаз, сердечно-сосудистой, дыхательной и других систем организма. Эту генетическую мутацию нельзя предотвратить или вылечить, но правильно подобранное лечение способно продлить пациентам жизнь и предупредить развитие опасных осложнений.
Причины синдрома Марфана
Данное генетическое заболевание вызвано дефектом гена FBN1 в длинном плече 15 хромосомы. Этот ген кодирует белок гликопротеин фибриллин-1, который отвечает за прочность и эластичность соединительной ткани. Соответственно, все проявления патологии связаны с тем, что соединительнотканные структуры в организме человека теряют свои нормальные свойства.
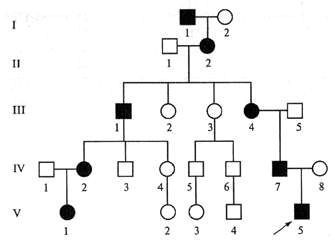
Наследуется мутация по аутосомно-доминантному признаку, то есть дети получают патологический ген от родителей, которые страдают от патологии. При этом шанс ребенка получить мутацию от одного из родителей составляет 50% (рис. 1). Синдром не передается через поколение: здоровые дети больных родителей не могут передать ген своим потомкам.
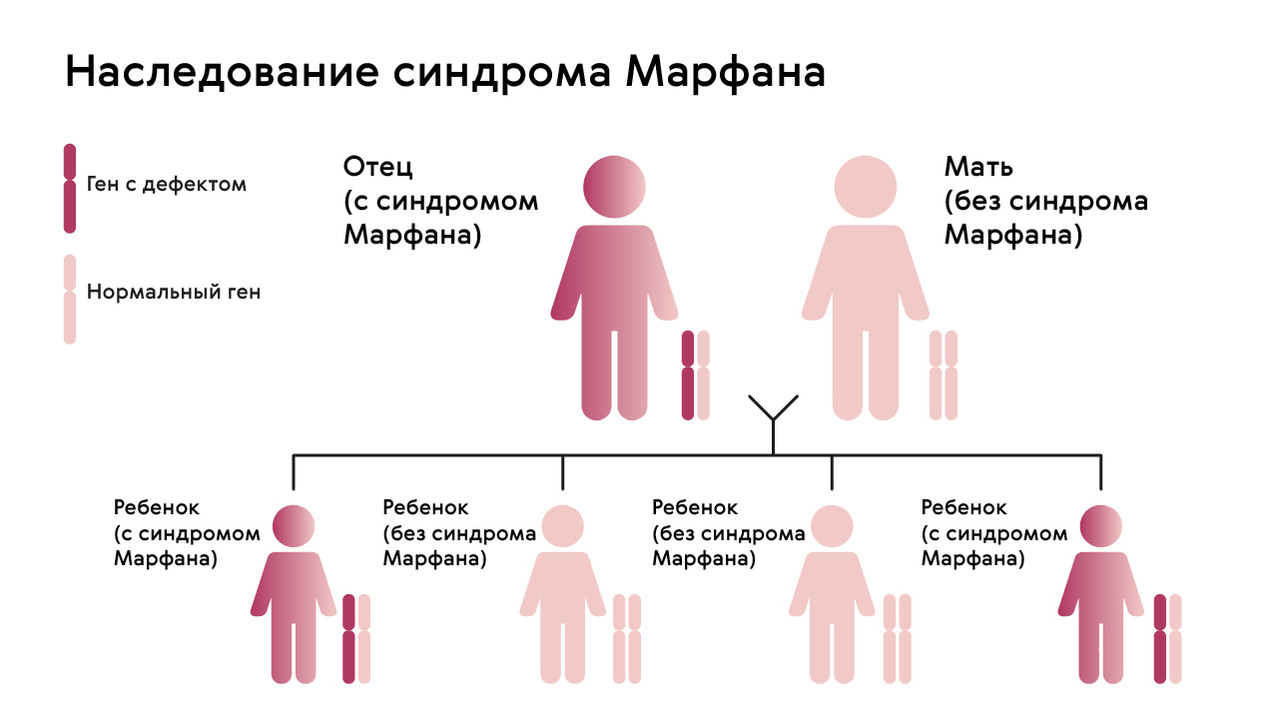
Рисунок. 1. Схема наследования синдрома Марфана. Источник: МедПортал
Однако примерно у 25% людей с синдромом Марфана никто из родителей не оказывается носителем аномалии гена FBN1: в таком случае мутация развивается спонтанно.
До сих пор не выявлено определенных факторов риска развития этого генетического нарушения: заболевание встречается одинаково часто среди мужчин и женщин, а его распространенность не зависит от расы или этнической группы. Частота заболеваемости у этой патологии составляет примерно 1 случай на 5–10 тысяч.
Если клинические признаки мутации ярко выражены, заподозрить болезнь можно уже в первые месяцы жизни ребенка, но стертые формы заболевания часто проявляются уже во взрослом возрасте, когда пациент обращается к врачам по поводу различных проявлений синдрома.
Важно! Не стоит записываться на генетическое обследование в качестве медосмотра. Поиски «поломки» гена FBN1 оправданы только в случае, если болезнь проявляет себя характерными признаками: бессимптомное носительство этой мутации невозможно. Если у одного из родителей установлен этот диагноз, будущей маме следует пройти генетическое обследование еще до родов. Это позволит заранее узнать, передалась ли аномалия ребенку.
Классификация синдрома Марфана
Выделяют несколько форм заболевания в зависимости от особенностей клинических проявлений генетической мутации.
Существуют две основные клинические формы патологии:
- Стертая. Таким пациентам «везет» больше: аномалия у них проявляется поражениями только одной-двух систем организма, а симптомы выражены незначительно. Люди могут жить практически нормальной жизнью, несмотря на болезнь.
- Выраженная. В таких случаях поражаются три и более систем организма, либо значительно нарушается функционирование одной из систем.
В зависимости от степени проявления выделяют легкие, среднетяжелые и тяжелые формы синдрома Марфана. Тяжелые патологии встречаются гораздо реже: частота их выявления составляет примерно 1 на 25–50 тысяч человек.
Принципиальную роль в определении прогноза болезни играет характер ее течения:
- Прогрессирующий. В этом случае постоянно появляются новые симптомы заболевания, степень тяжести увеличивается, а с каждым годом жизни пациента возрастают риски фатальных осложнений.
- Стабильный. Такой характер считается наиболее благоприятным: у пациентов со стабильными проявлениями синдрома Марфана клиническая картина практически не меняется на протяжении жизни.
Выделяют три разных, но похожих заболевания:
- Синдром Марфана — стертая форма патологии с положительным результатом генетического тестирования.
- Болезнь Марфана — классическая клиническая картина с подтвержденным семейным наследованием.
- Марфаноподобный синдром — проявление патологии соединительной ткани без генетической мутации.
Первые признаки заболевания чаще всего проявляются еще в детском возрасте. К подростковому периоду становится понятно, насколько быстро у пациента прогрессирует болезнь, вызванная мутацией гена FBN1.
Симптомы синдрома Марфана
Проявления генетического дефекта могут быть выражены в разной степени: от легкого изменения строения соединительной ткани до тяжелых нарушений жизненно важных функций организма. Иными словами, внешние признаки аномалии у разных пациентов могут значительно отличаться, несмотря на одинаковый генетический дефект.
Классической триадой синдрома Марфана считаются: скелетные нарушения, смещение хрусталика и расслоение аорты (рис. 2). Также системное поражение соединительной ткани у пациентов становится причиной развития нарушений работы практически всех органов и систем организма.
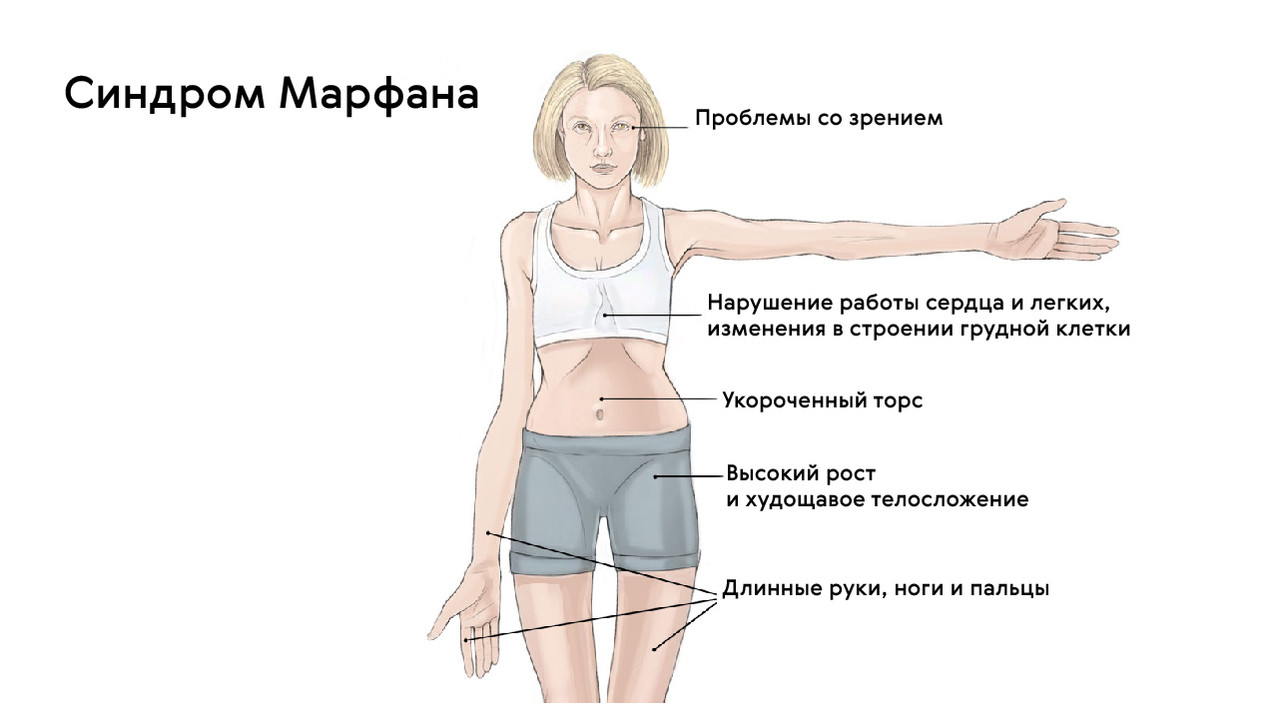
Рисунок 2. Классические проявления синдрома Марфана. Источник: zinkmd.com
Костно-мышечная система
Выраженность симптомов поражения опорно-двигательного аппарата зависит от тяжести случая и особенностей организма пациента.
Для людей с синдромом Марфана характерен чрезвычайно высокий рост: обычно дети «перерастают» всех членов семьи. При этом часто, особенно в детском возрасте, привлекает внимание нестандартная длина рук: их размах оказывается больше, чем длина тела.
Яркий симптом болезни — патологически удлиненные и тонкие пальцы, так называемые «пальцы паука» (арахнодактилия) (рис. 3).
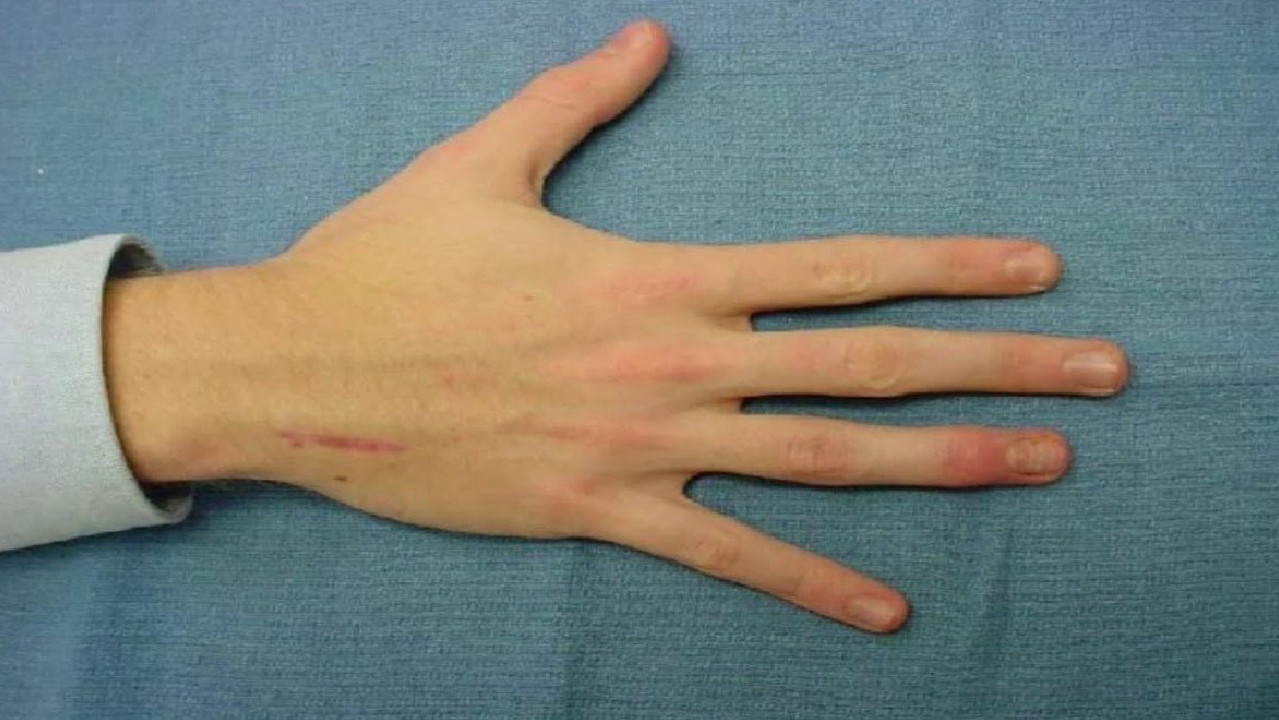
Рисунок 3. Арахнодактилия. Источник: twitter.com
Проверить наличие симптома можно с помощью теста большого пальца кисти — у пациентов с арахнодактилией часть большого пальца (дистальная фаланга) выступает за край сжатой в кулак ладони (рис. 4).
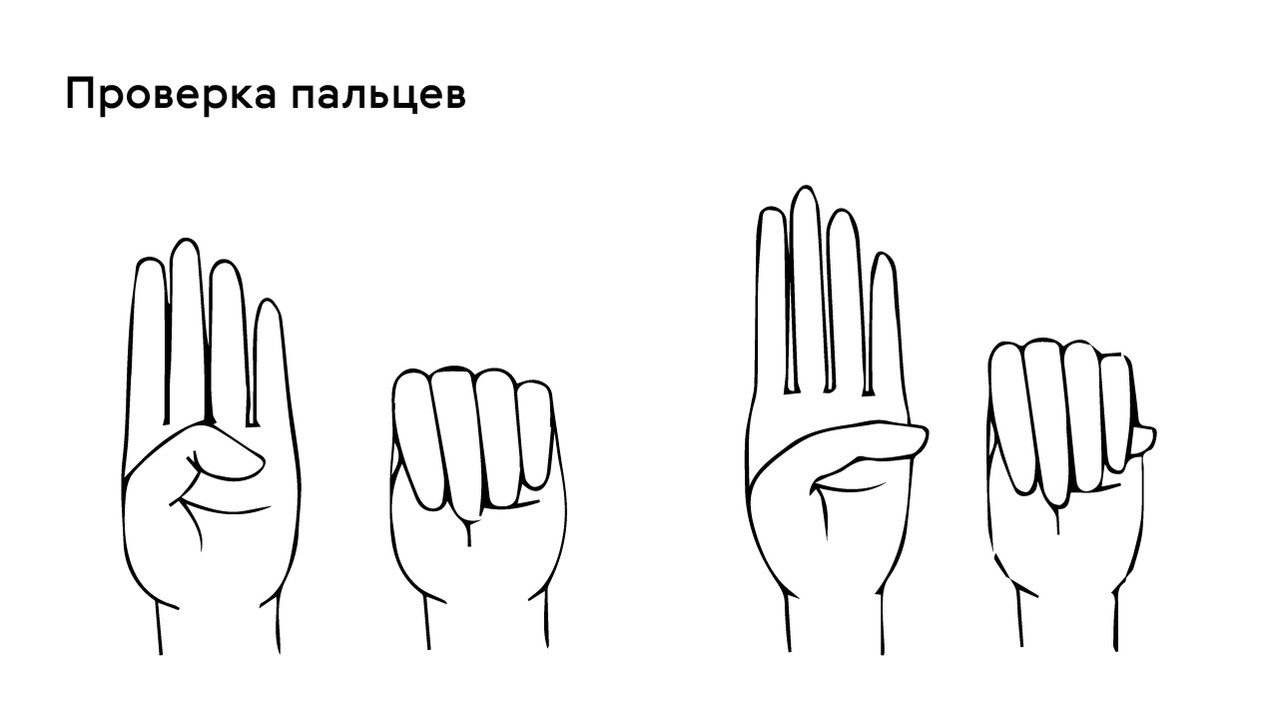
Рисунок 4. Проверка на арахнодактилию. Источник
Лицо людей с синдромом Марфана обычно вытянутое и худое. Этому способствует высокое положение свода верхнего неба, удлиненный череп и патологическая худоба.
Также для таких пациентов характерны деформации грудной клетки, которые могут быть в двух вариантах: смещение грудины внутрь (воронкообразная грудь) или наружу (килевидная грудь, рис. 5).
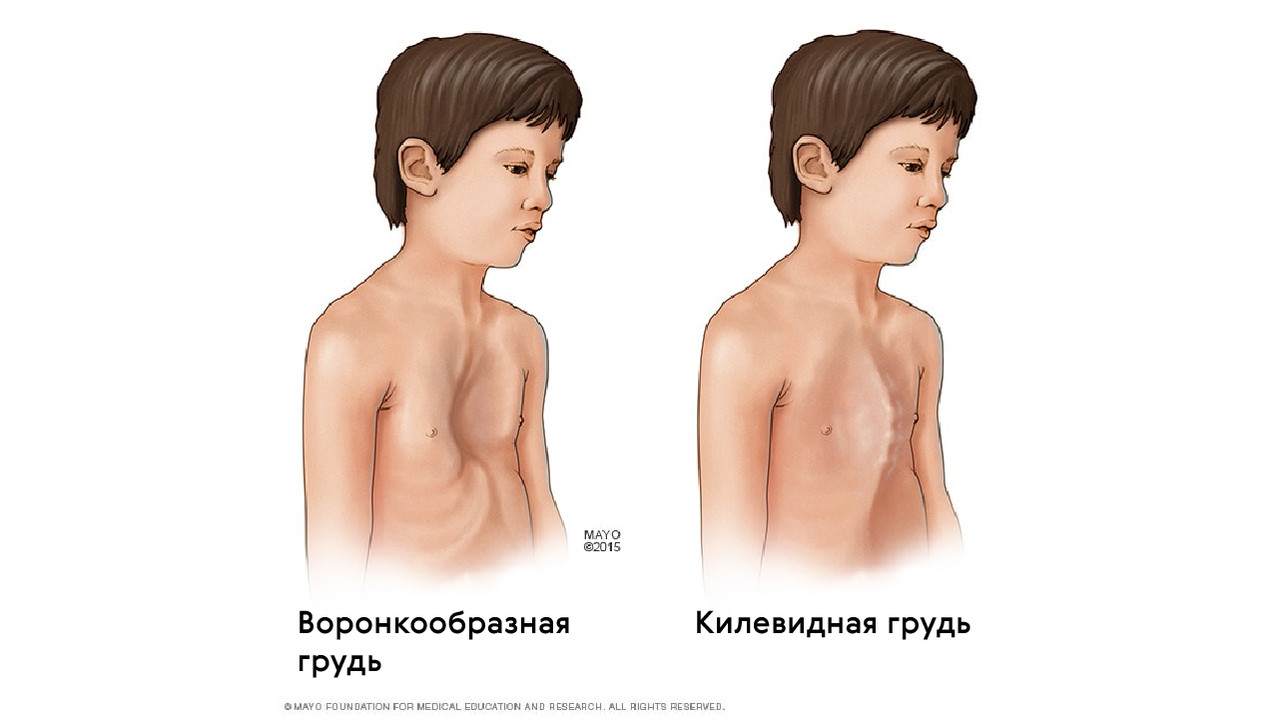
Рисунок 5. Воронкообразная и килевидная деформации грудной клетки при синдроме Марфана. Источник: mayoclinic.org
Осанка пациентов с синдромом Марфана в большинстве случаев нарушена. Чаще всего определяются различные степени выраженности сколиоза (отклонение позвоночного столба в сторону) или кифоза (формирование «горба»).
Кроме того, пациенты с FBN1 мутацией часто страдают от:
- плоскостопия;
- повышенной подвижности всех суставов;
- слабости связочного аппарата.
У пациентов с синдромом Марфана часто плохо развиты мышечные структуры и практически нет подкожно-жирового слоя. Движения пациентов с этой патологией неловкие, они часто получают различные травмы.
Высокий темп роста и нарушения выработки белков соединительной ткани определяют патологии кожи у людей с мутацией гена FBN1. Клинически это проявляется в виде повышенной растяжимости кожных структур с образованием светлых полос — «растяжек» (стрий).
Зрение
Дефекты гена FBN1 определяют склонность к патологиям зрительной системы. Чаще всего повреждения глаз у пациентов с синдромом Марфана включают в себя:
- выраженную близорукость;
- подвывих или изменение положения хрусталика;
- высокий риск внезапной отслойки сетчатки глаза.
Кроме того, у таких пациентов гораздо раньше может развиться катаракта или глаукома: те патологии органа зрения, которые считаются возрастными у здоровых людей.
Органы дыхания
В легких пациентов с синдромом Марфана может патологически разрастаться соединительная ткань. Это приводит к формированию сужения бронхов и легочного фиброза. Нередко на фоне генетической мутации развивается бронхиальная астма или хроническое обструктивное заболевание легких. Генетическая аномалия также определяет возможность развития спонтанного пневмоторакса — неотложной ситуации, в которой в полость вокруг легких попадает воздух, и легкое резко уменьшается в размерах («спадается»).
Желудочно-кишечный тракт
Процессы пищеварения у людей с FBN1 мутацией меняются: нарушается моторика кишечника, появляются патологии желчного пузыря, часто развиваются гастриты, язвенные дефекты, дисбиоз.
Почечный аппарат
У пациентов с синдромом Марфана чаще находят аномалии почек: опущение органов, расширение почечных лоханок, патологическую подвижность почек.
Нервная система и психическая сфера
Хотя в большинстве случаев у пациентов с синдромом Марфана не происходит нарушений работы мозговых структур, некоторые патологические изменения нервной системы могут присутствовать. Например, расширение соединительнотканной капсулы, которая окружает спинной мозг, может приводить к нарушениям движений в нижних конечностях, работы мочевого пузыря и кишечника. Для таких пациентов характерно развитие синдрома хронической усталости — астения, склонность к депрессии. Интеллектуальная деятельность в большинстве случаев не нарушена, даже наоборот: среди пациентов с синдромом Марфана есть люди с интеллектом значительно выше среднего.
Сердечно-сосудистая система
Кардиологи выявляют нарушения ритма сердца у людей с синдромом Марфана. У пациентов с этой патологией часто нарушается структура аортального клапана — соединительнотканной перегородки, которая предупреждает обратный ток крови из аорты в сердце. Это приводит к развитию порока сердца — аортальной недостаточности. Также могут развиваться другие пороки сердца, например, пролапс или недостаточность митрального клапана, а на пораженных участках часто развивается инфекционно-воспалительный процесс — бактериальный эндокардит.
Самую большую опасность представляют патологические изменения в главном сосуде организма — аорте. У 65–100% людей с синдромом Марфана есть большой риск поражения луковицы (наиболее близкая к сердцу часть аорты) и восходящей дуги этой артерии — тех частей, которые непосредственно выходят из сердца. Поскольку внутренний слой стенки сосудов также содержит волокна соединительной ткани, они склонны к износу, а давление крови в аорте выше, чем в других участках сосудистого русла. Это приводит к тому, что сосуд постепенно расширяется, и может произойти патологическое скопление крови между сосудистыми стенками с формированием мешковидного выпячивания (аневризмы) или спонтанный разрыв артерии.
Почему при определении признаков синдрома Марфана нужно обратиться к врачу?
Сама по себе генетическая аномалия совместима с жизнью. Однако опасны последствия болезни, вызванной FBN1 мутацией:
- разрывы крупных сосудов, чаще всего — аорты;
- хроническая сердечная недостаточность — неспособность сердца обеспечивать необходимую работу для кровоснабжения всех органов;
- снижение остроты зрения или полная потеря зрительной функции.
Разрыв аневризмы аорты или другого магистрального сосуда часто заканчивается моментальным летальным исходом. Хроническая сердечная недостаточность может перейти в острую форму, а без экстренной медицинской помощи также привести к фатальным последствиям — внезапной коронарной смерти. Именно эти осложнения чаще всего приводит к гибели детей с синдромом Марфана. Особая опасность ждет женщину с синдромом мутации гена FBN1 во время беременности: повышенная нагрузка на аорту в разы увеличивает риск ее разрыва.
Чтобы предупредить развитие опасных осложнений и компенсировать возникающие нарушения, родителям нужно как можно раньше обратиться за медицинской помощью при первом подозрении на синдром Марфана у ребенка. При этом важно не только однократно провести обследование, но и стать на учет к врачам, которые занимаются коррекцией проявлений синдрома:
- специалисту по генетическим болезням;
- кардиологу;
- ортопеду-вертебрологу;
- дерматологу;
- офтальмологу;
- гастроэнтерологу.
Список специалистов зависит от степени выраженности заболевания, при этом регулярно необходимо проходить комплексные профилактические осмотры для раннего выявления новых нарушений.
Синдром Марфана — болезнь гениев?
С синдромом Марфана связаны не только многочисленные поводы для обращения к врачам. Часто люди с мутацией гена FBN1 компенсируют физические проявления болезни интеллектуальными способностями, поэтому это генетическое заболевание даже называют «синдромом гениев». Считается, что повышенный выброс адреналина из-за патологических изменений в надпочечниках определяет высокий тонус умственной и психической активности у таких пациентов. Именно поэтому в числе людей с синдромом Марфана можно найти известных личностей. Например, Юлию Цезарю, Аврааму Линкольну и Шарлю де Голлю патология не помешала стать известными политическими деятелями; Ганс Христиан Андерсен и Корней Чуковский создали уникальные литературные произведения, а Никколо Паганини прославился как гениальный музыкант.
Современные знаменитости также не скрывают свои недостатки и становятся еще более популярными из-за генетического дефекта. Например, солисту американской рок-группы Deerhunter Брэдфорду Коксу нетипичная внешность придает особый шарм, а испанский актер Хавьер Ботет очень востребован, поскольку правдоподобно и талантливо играет отрицательных героев в голливудских фильмах ужасов (рис. 6).

Рисунок 6. Актер Хавьер Ботет, страдающий синдромом Марфана. Источник: kinopoisk.ru
Диагностика синдрома Марфана
Диагностика генетической аномалии включает в себя комплекс мероприятий по определению всех симптомов болезни, а также изучению вероятности развития мутации:
- Сбор жалоб — детальное изучение всех патологических признаков.
- Определение анамнеза — выяснение состояния здоровья родителей.
- Тщательный осмотр, измерение роста, размаха рук и других антропометрических показателей. Скрининговый тест для детей в возрасте 7–18 лет — это измерение длины среднего пальца руки. У пациентов с синдромом Марфана показатель превышает отметку в 10 см.
Генетическое обследование включает в себя выявление генотипа ДНК — идентификацию мутаций в гене FBN1. При возможности назначают специфические лабораторные тесты — определение выведения с мочой метаболитов соединительной ткани, таких как оксипролин и гликозаминогликаны.
Чтобы подтвердить нарушения развития соединительной ткани и оценить степень выраженности мутации гена FBN1, пациентам с подозрением на синдром Марфана назначают:
- ЭКГ;
- УЗИ сердца;
- КТ-ангиографию аорты и других сосудов;
- КТ грудной и брюшной полостей;
- МРТ позвоночника и головного мозга;
- специфические обследования на осмотре у офтальмолога;
- биопсию кожи.
Для окончательного определения диагноза используют общепринятые Гентские критерии 2010 года, согласно которым диагноз устанавливают в случаях:
- подтвержденной мутации гена FBN1 и расширения корня аорты или эктопией хрусталика;
- подтвержденного расширения корня аорты в сочетании с эктопией хрусталика;
- подтвержденной эктопии хрусталика в сочетании с любыми признаками системного поражения соединительной ткани.
Важно! Существует группа «марфаноподобных» синдромов, при которых внешне пациенты очень напоминают больных с аномалией гена FBN1, но причина их патологии скрывается в других нарушениях. К примеру, гомоцистинурия — это обменное заболевание, которое проявляется системными изменениями соединительной ткани, но может приводить к внезапным инсультам и существенно замедляет умственное развитие ребенка. Поэтому важно точно определить причину заболевания соединительной ткани и своевременно начать лечение.
Лечение синдрома Марфана
К сожалению, на сегодняшний день лекарственные методы терапии этой генетической патологии еще не разработаны. Однако пациентам с синдромом Марфана важно соблюдать все назначения врачей, чтобы устранить симптомы патологии и замедлить темпы ее развития.
Лечение зависит от клинических проявлений болезни:
- при аневризме аорты назначают препараты, которые снижают частоту и силу сердечных сокращений, снимая избыточную нагрузку на сосуды;
- пациентам с синдромом Марфана часто назначают антигипертензивные препараты для снижения артериального давления;
- хондроитин и глюкозамин относятся к естественным компонентам соединительной ткани — их прием улучшает структуру хрящей и предупреждает патологии суставов;
- для стимуляции образования коллагена выписывают специальные БАДы — L-карнитин, витамины из групп С, D, Е, В, а также кальций, цинк и другие пищевые добавки.
Пациентам противопоказаны физические нагрузки, постоянная активность, травмоопасные игры. Рацион питания людей с синдромом Марфана должен быть насыщен белками, полезными жирными кислотами, микро- и макроэлементами. Для поддержки структур скелета пациентам с мутацией в гене FBN1 показано ношение корсетов, укрепление мышц с помощью ЛФК и оздоровительного массажа.
В некоторых случаях может помочь только хирургическое лечение — операции по замене части аорты, клапанов, исправлению костных патологий или коррекции патологий глаза, которые существенно снижают риски опасных осложнений.
Прогноз
Современные методы исследования в медицине позволяют выявлять заболевание у детей в раннем возрасте. Это помогает повысить качество жизни таких пациентов и предупредить раннюю смертность. Продолжительность жизни людей с синдромом Марфана при бережном отношении к своему здоровью достигает 70 лет. Прогноз болезни во многом зависит от выраженности сердечно-сосудистых патологий, поскольку выживание пациентов с этой генетической аномалией определяет состояние аорты и риск ее спонтанного разрыва. Такие люди требуют постоянного наблюдения у врачей различных специальностей для своевременной коррекции проявлений синдрома.
Заключение
Конечно, жизнь с этой генетической мутацией становится сложнее, но при правильном подходе к собственному здоровью и своевременному обследованию у врачей пациентам с синдромом Марфана удается компенсировать все проявления заболевания и не допустить развития фатальных осложнений.
Активисты с синдромом Марфана создают тематические сообщества по всему миру: мощная поддержка людей с такой же генетической аномалией позволяет пациентам не чувствовать себя одинокими.
Синдром Марфана
Информацию из данного раздела нельзя использовать для самодиагностики и самолечения. В случае боли или иного обострения заболевания диагностические исследования должен назначать только лечащий врач. Для постановки диагноза и правильного назначения лечения следует обращаться к Вашему лечащему врачу.
Синдром Марфана: причины появления, симптомы, диагностика и способы лечения.
Определение
Синдромом Марфана, или Марфана-Ашара, называют наследственное заболевание соединительной ткани с преимущественным поражением сердечнососудистой системы, скелета и органа зрения. Частота синдрома в популяции составляет от 1:3000 до 1:15000. Впервые его описали французские врачи Антонин Бернард Марфан (1896) и Эмиль Шарль Ашар (1902).
Причины появления синдрома Марфана
Синдром Марфана имеет аутосомно-доминантный тип наследования (то есть дети получают патологический ген от родителей, которые страдают данным заболеванием). Молекулярной основой синдрома Марфана является нарушение синтеза одного из белков соединительной ткани – фибриллина, который в норме придает ей эластичность и сократительную способность.
При СМ наблюдается дефицит фибриллина или его аномальное строение, поэтому соединительная ткань обладает повышенной растяжимостью и теряет способность выдерживать физиологические нагрузки.
Классификация заболевания
В зависимости от количества пораженных систем организма выделяют несколько форм синдрома Марфана:
- стертую – со слабо выраженными изменениями в 1-2 системах;
- выраженную – со слабо выраженными изменениями в 3 системах; значительно выраженными изменениями хотя бы в 1 системе; выраженными изменениями в 2-3 и более системах.
Принципиальную роль в определении прогноза болезни играет характер ее течения.
Прогрессирующий. В этом случае постоянно появляются новые симптомы заболевания, степень тяжести увеличивается, с каждым годом жизни пациента возрастают риски фатальных осложнений.
Стабильный. Такой характер считается наиболее благоприятным: у пациентов со стабильными проявлениями синдрома Марфана клиническая картина практически не меняется на протяжении жизни.
Симптомы синдрома Марфана
Поскольку фибриллин находится в соединительной ткани различных органов, симптоматика СМ весьма разнообразна.
Симптомы патологии сердечно-сосудистой системы
- Изменения сердечно-сосудистой системы отмечаются у большинства больных. Их основная причина – потеря способности стенок артерий и клапанных структур сердца выдерживать естественные гемодинамические нагрузки. Наиболее частая сердечная патология при синдроме Марфана –недостаточность митрального клапана, когда наблюдается поражение эластических структур створок и сухожильных нитей клапана с развитием его дисфункции. Со временем у многих больных эта дисфункция переходит в умеренную или тяжелую митральную недостаточность, требующую хирургической коррекции. Реже можно наблюдать аортальную и трикуспидальную недостаточность. Стенозы клапанов для СМ не характерны. При небольшой или умеренной хронической митральной недостаточности жалобы обычно отсутствуют (особенно при медленном нарастании недостаточности). Со временем появляются одышка, ощущение быстрой утомляемости и учащенное сердцебиение.
Клапанные пороки нередко осложняются инфекционным эндокардитом (воспалением внутренней оболочки сердца) – внезапно повышается температура, появляются озноб, боль в суставах, бледность кожных покровов и слизистых).
Кроме того, формируется геморрагическая сыпь, деформируются фаланги пальцев и ногтевые пластины (симптом «барабанных палочек» и «часовых стекол»).
Самую большую опасность представляют, пожалуй, патологические изменения аорты. Поскольку внутренний слой стенки сосудов также состоит из волокон соединительной ткани, сосуды постепенно изнашиваются. Принимая во внимание тот факт, что давление крови в аорте выше, чем на других участках сосудистого русла, это приводит к постепенному ее расширению, патологическому скоплению крови между сосудистыми стенками и формированию аневризмы или спонтанному разрыву. Основные симптомы аневризмы аорты следующие: осиплость голоса; нехватка воздуха; боль в плече; кровохарканье, боль в спине.
Симптомы поражения опорно-двигательного аппарата
Выраженность симптомов поражения опорно-двигательного аппарата зависит от тяжести заболевания и особенностей организма пациента. Люди с синдромом Марфана обычно отличаются высоким ростом. У детей с СМ очень длинные, непропорциональные росту руки и патологически удлиненные, тонкие пальцы, так называемые пальцы паука (арахнодактилия).
Лицо у людей с синдромом Марфана обычно вытянутое и худое. Этому способствует высокое положение свода верхнего неба, удлиненный череп и патологическая худоба.
Для пациентов характерны деформации грудной клетки, которые могут быть двух вариантов: смещение грудины внутрь (воронкообразная грудь) или наружу (килевидная грудь).
Часто определяются различной степени выраженности сколиоз (отклонения позвоночного столба в сторону) или кифоз (формирование горба).
Кроме того, у пациентов с СМ нередко диагностируют плоскостопие, повышенную подвижность суставов, слабость связочного аппарата, неразвитость мышечных структур и подкожно-жирового слоя.

Симптомы поражения кожи
Высокий темп роста и нарушение выработки белков соединительной ткани определяют патологии кожи у людей с мутацией гена FBN1. Клинически это проявляется повышенной растяжимостью кожного покрова с образованием растяжек (стрий).
Симптомы поражения органа зрения
Чаще всего повреждения глаз у пациентов с синдромом Марфана включают:
- выраженную близорукость;
- подвывих или изменение положения хрусталика;
- высокий риск внезапной отслойки сетчатки глаза.
Симптомы поражения органов дыхания
В легких пациентов с СМ может разрастаться может патологически разрастаться соединительная ткань, приводя к сужению бронхов и легочному фиброзу. Нередко на фоне генетической мутации манифестирует бронхиальная астма или хроническое обструктивное заболевание легких. Генетическая аномалия также определяет риск развития спонтанного пневмоторакса — неотложной ситуации, при которой в плевральную полость попадает воздух, и легкое спадается: у пациента внезапно появляется одышка, резкая или ноющая боль в груди, сухой кашель.
Симптомы поражения желудочно-кишечного тракта
У людей с FBN1 мутацией нарушается моторика кишечника, появляются патологии желчного пузыря, часто развиваются гастриты, язвенные дефекты, дисбиоз. Пациент испытывает привкус горечи во рту, тяжесть в области правого подреберья, в надчревной области. Часто беспокоит изжога, отрыжка, вздутие живота.
Симптомы поражения мочевыделительной системы
У пациентов с синдромом Марфана чаще находят аномалии почек: опущение органов, расширение почечных лоханок, патологическую подвижность почек.
Признаком нефроптоза легкой степени являются периодические одно- или двусторонние боли в проекции почек. Как правило, они проходят после непродолжительного пребывания в горизонтальном положении. Боль обычно тупая, ноющая, слабая или умеренная. Усугубление опущения ведет к нарушению оттока мочи и создает условия для развития инфекционных процессов и гидронефроза (скопления жидкости в почке). При присоединении инфекции может повышаться температура тела, появляться слизь и гной в моче.
По мере снижения функции почек у больного развиваются отеки, гипертензия и другие системные нарушения.
Симптомы поражения нервной системы
Расширение соединительнотканной капсулы, которая окружает спинной мозг, может приводить к нарушениям движений в нижних конечностях, работы мочевого пузыря и кишечника. У таких пациентов развивается синдром хронической усталости, проявляющийся астенией и депрессивными расстройствами.
Интеллектуальная деятельность в большинстве случаев не нарушена, наоборот – среди пациентов с синдромом Марфана есть люди с интеллектом значительно выше среднего.
Диагностика синдрома Марфана
Диагностика генетической аномалии включает комплекс мероприятий по определению всех симптомов болезни, а также изучению вероятности развития мутации:
- Сбор жалоб — детальное изучение всех патологических признаков.
- Определение анамнеза — выяснение состояния здоровья родителей.
- Тщательный осмотр, измерение роста, размаха рук и других антропометрических показателей. Скрининговый тест для детей в возрасте 7–18 лет — измерение длины среднего пальца руки. У пациентов с синдромом Марфана показатель превышает отметку 10 см.
- Лабораторное обследование.
Для подтверждения нарушений развития соединительной ткани и оценки степени выраженности мутации гена FBN1 пациентам с подозрением на синдром Марфана назначают:
Электрокардиография (ЭКГ) – повсеместно распространенный метод изучения работы сердца, в основе которого лежит графическое изображение электрических импульсов сердца.
Синдром Марфана

Синдром Марфана — это заболевание соединительной ткани, вызванное наследственным дефектом в гене FBN1 на хромосоме 15, который кодирует белок фибриллин. Подобное нарушение оказывает влияние на работу всех органов, но особенно страдают скелетно-мышечная, сердечно-сосудистая системы и зрительный аппарат.
Диагноз синдрома Марфана основывается на семейном анамнезе, физическом осмотре, наличии определенных диагностических критериев, молекулярно-генетическом тестировании и пр. Важно прохождение регулярного обследования для своевременного выявления факторов риска для жизни. Лечение направлено на корректирование дефектов, предотвращение осложнений, поддержку жизнедеятельности.
Рассказывает специалист ЦМРТ
Дата публикации: 02 Августа 2021 года
Дата проверки: 30 Ноября 2021 года
Содержание статьи
Причины синдрома Марфана
Болезнь обнаруживают у 1 из 10 000 человек. В основе патогенеза синдрома Марфана — дефект гена на хромосоме 15, который определяет структуру фибриллина. Данный белок является основным компонентом микрофибрилл, связанных с эластином. Последние обнаруживают в большинстве соединительных тканей, крупных кровеносных сосудах и поддерживающих хрусталик связках.
Болезнь наследуется по аутосомно-доминантному типу: если один из родителей болен, вероятность развития патологии у ребенка составляет 50%. Зарегистрированы случаи, когда у здоровой пары рождается ребенок с подобной генетической аномалией: спонтанная мутация встречается в 1 из 4 случаев.
Как проявляется синдром Марфана
Степень выраженности дефектов органов и систем индивидуальна. Некоторые пациенты могут иметь частичную форму марфаноподобного синдрома с легкими, практически незаметными проявлениями, другие – страдают более серьезно. Огромный фенотипический спектр объясняют разнообразием мутаций в гене и изменениями в других структурных единицах наследственности, со сходными проявлениями. Отклонение внешнего вида от нормы иногда можно заметить в детском возрасте. Повсеместное присутствие соединительной ткани приводит к разнообразию симптомов.
Признаки синдрома Марфана:
- килевидное или воронкообразное искривления грудной клетки, деформация позвоночника
- непропорциональное туловище, высокий рост
- длинные (паучьи) пальцы (при положительном знаке запястья большой и указательный пальцы при обхвате второй руки перекрывают друг друга)
- увеличение амплитуды движений в конечностях
- патологическая гибкость суставов
- плоско-вальгусная деформация стопы
- дефицит веса
- арочное небо, деформация зубного ряда
- нетипичное лицо (долихоцефалия, недоразвитие скуловой кости, впалые глаза
- зрительная дисфункция на фоне эктопии (вывиха) хрусталика, аномального уплощения роговицы, миопии и пр
- одышка, слабость
- кожные стрии (растяжки)
- рецидивирующие грыжи и пр.
Классификация синдрома Марфана
- стертая форму болезни — в процесс вовлечено не более двух систем органов, проявления минимальны
- выраженная, для которой характерны незначительные изменения в работе трех систем или присутствуют клинически значимые симптомы, связанные с серьезным поражением одной и более систем.
Болезнь может носить стабильный или прогрессирующий характер.
Как диагностировать
Алгоритм обследования зависит от вовлеченных систем. Обязательны консультации специалистов: генетика, офтальмолога, кардиолога, пульмонолога, гастроэнтеролога.
Инструментальное обследование опирается на:
- Рентгенографию или компьютерное сканирование кистей рук, стоп, грудной клетки, позвоночника, костей тазобедренных суставов, черепа.
- МРТ — если требуется диагностика изменений внутренних органов: аномалий развития кишечника, пороков сердца, почек и пр. Торакоабдоминальная магнитно-резонансная ангиография обеспечивает полную оценку артериальной системы.
- ЭКГ, ультразвуковое сканирование, трансторакальную эхокардиографию (ЭхоКГ) сердца и сосудов;
К какому врачу обратиться
Пациенты приходят на прием нужного специалиста с преобладающими симптомами. Три “главных” врача для больных с марфаноподобным синдромом: кардиолог, окулист, хирург-ортопед.
Как лечится синдром Марфана?
Специфического лечения синдрома Марфана не существует. Медикаментозная терапия бета-блокаторами направлена на предотвращение развития сердечно-сосудистых осложнений — дилатации и расслаивающей аневризмы аорты, поражения митрального и аортального клапанов. Дополнительно пациентам рекомендуют прием препаратов, улучшающих метаболизм.
Основным показанием к операции на опорно-двигательном аппарате является прогрессирование сколиоза от средней до тяжелой степени. Деформация грудной клетки может влиять на сердечно-легочную механику; дефекты можно исправить оперативным путем. Появление успешной реконструктивной хирургии аневризм корня аорты, замены аортального и митрального клапанов изменили общие долгосрочные перспективы для пациентов с этим заболеванием.
Зрение корректируется ношением очков, линз, имплантацией искусственного хрусталика, выполнением лазерных операций.
Последствия
Без обращения за специализированной помощью ожидаемы:
- со стороны сердечно-сосудистой системы – разрыв аневризмы, аритмии (фибрилляции), декомпенсированная сердечная недостаточность, износ клапанного аппарата
- слепота в результате нелеченной и нераспознанной глаукомы, отслойки сетчатки, катаракты
- тромбозы
- рестриктивные заболевания дыхательных путей, формирование легочного сердца из-за деформаций позвоночника
Профилактика
Профилактических мероприятий не разработано. Превентивные меры включают:
- медико-генетическое консультирование при имеющихся в роду близких родственников с болезнью Марфана
- пренатальную диагностику
Лечение синдрома Марфана в клиниках ЦМРТ
При ранней диагностике и профилактическом лечении ожидаемая продолжительность жизни аналогична средней в популяции. В медицинских центрах ЦМРТ пациент может пройти обследование на оборудовании экспертного класса – МРТ, КТ, ЭКГ, ЭхоКГ и пр., получить консультацию специалиста – кардиолога, невролога, ортопеда-травматолога; подобрать ортезы при диспропорции тела; заниматься лечебной физкультурой под руководством инструктора, реабилитироваться с помощью массажа, физиотерапии.
Источники
Российское научное медицинское общество терапевтов (РНМОТ) Клинические рекомендации: Дисплазии соединительной ткани, 2017 г.
Пересмотренные Гентские критерии диагностики синдрома Марфана THE REVISED GHENT NOSOLOGY FOR THE MARFAN SYNDROME B.L. Loeys, H.C. Dietz, A.C . Braverman, B.L. Callewaert, J. De Backer, R.B. Devereux, Y. Hilhorst-Hofstee, G. Jondeau, L. Faivre, D.M. Milewicz, Pyeritz R.E., P.D. Sponseller, P. Wordsworth, A. M. De Paepe

