
Синдром Ретта у девочек, детей. Что это такое, симптомы, описание, лечение, прогноз, причины
Синдром Ретта является необычным нарушением развития мозга, с напоминающими аутизм или нейродегенеративное заболевание признаками, но в действительности не является ни одним из них. Синдром Ретта представляет собой характерный комплекс клинических проявлений, включающих раннюю психомоторную регрессию с аутистическими проявлениями, замещение целенаправленных действий рук стереотипными движениями, атаксией и апраксией при ходьбе и приобретенной микроцефалией (Hagberg, 1989). В таблице 4.4 представлены международные критерии диагностики (Rett Syndrome Diagnostic Criteria Work Group, 1988; Trevathan и Naidu, 1988). За редким исключением данное заболевание встречается только у девочек (Zoghbi, 1988, Hagberg, 1989).
а) Распространенность. Распространенность синдрома Ретта в Швеции и западной Шотландии, составляет 1 на 10000 и 1 на 18000 девочек (Kerr и Stephenson, 1985; Hagberg, 1993; Bienvenu et al., 2006).
б) Патогенез. Синдром Ретта приблизительно в 80% случаев связан с мутациями гена МЕСР2, и длительное время считалось, что мутация данного гена является причиной синдрома Ретта. Тем не менее, фенотипические проявления мутаций гена МЕСР2 разнообразны и включают задержку умственного развития с припадками или без них, фенотип, подобный синдрому Ангельмана, и аутизм (Zoghbi, 2005). Как минимум еще один ген (CDKL5) связан с развитием судорожного варианта заболевания. Ген МЕСР2 является ингибитором фактора транскрипции, способным отключать несколько важных для развития головного мозга генов, а в связи с экспрессированием в различных типах клеток и органов— влиять на соматическое развитие в целом. В этой связи, представляется вероятным, что мутация гена МЕСР2 при синдроме Ретта является частью последовательной цепи событий, приводящих к развитию ряда сцепленных с Х-хромосомой нарушений развития нервной системы.
В настоящее время синдром Ретта представляется скорее патологией развития, а не дегенеративным заболеванием (Naidu, 1997). Структурные аномалии мозга выражены слабо и включают маленький размер мозга с плотно расположенными нейронами и снижением клеточных процессов. В подавляющем большинстве случаев заболевание не имеет наследственного характера, несмотря на то, что выборочное поражение девочек предполагает генетическое происхождение синдрома.

в) Клинические проявления. Течение синдрома Ретта имеет необычный характер. Заболевание начинается как прогрессирующее состояние с более или менее стремительной деградацией с утратой ранее полученных навыков.
Течение заболевания может быть разделено на четыре стадии. Дебют клинических симптомов приходится на возраст от шести месяцев до трех лет, в большинстве случаев заболевание манифестирует до 18 месяцев. Изначально развитие ребенка может не отличаться от нормы, но у больных девочек часто с рождения отмечается гипотония и слегка замедленное развитие (Einspieler et al., 2005).
Большинство девочек с синдромом Ретта развиваются нормально или почти нормально до 6-16 месяцев. Ретроспективно часто отмечается умеренная гипотония и минимальная задержка развития. Освоение целенаправленных движений рук является предварительным условием постановки диагноза. В дальнейшем у многих пациентов отмечается остановка развития или резкая утрата навыков (возможна потеря социальной улыбки, способности к взаимодействию и некоторых речевых навыков). Основным проявлением является утрата мануальных навыков. Данное проявление часто развивается стремительно в течение нескольких недель или носит «взрывной» характер, возникая в течение нескольких дней. Некоторые, но не все, дети становятся отчужденными, эмоционально отстраненными и описываются как «аутисты». У других медленно развивается малоэмоциональный стиль социального взаимодействия, иногда определяемый как аутистический.
Небольшое количество пациентов страдает от приступов ярости, тревоги, смущения и беспорядочной гиперактивности. При наличии аутистической или подобной аутистической фазы данное состояние может длиться от одного месяца до нескольких лет. Обычно к достижению школьного возраста (или не позднее пубертатного периода) аутистические симптомы начинают убывать, но не во всех случаях. По имеющимся данным, у большинства аутистов, вне зависимости от причины заболевания, присутствует одинаковый характерный тип развития.
Для девочек с синдромом Ретта характерны различные виды стереотипных движений рук, большая часть которых включают «движения в области средней линии», то есть обе руки «моются» или складываются по средней линии, обе руки засовываются в рот или шлепают по средней линии лба или шеи. На ранней стадии возможны более типичные для аутизма стереотипии с хлопаньем в ладоши.
У некоторых больных девочек зарегистрированы случаи смеха в середине ночи. Данное проявление возникает в результате нейрометаболических/неврологических нарушений в головном мозге (например, при мукополисахаридозе Санфилиппо) и встречается у многих девочек с аутизмом, не страдающих синдромом Ретта.
Бруксизм и гипервентиляция являются типичными проявлениями синдрома Ретта и иногда интерпретируются как признаки чрезвычайной тревожности, что не подтверждается опытом.
Третья стадия заболевания характеризуется медленным появлением неврологических признаков, таких как пирамидные знаки. Эпилептические припадки на данной стадии развиваются у 2/3-3/4 больных. Нередки предшествующие изменения ЭЭГ, включающие ритмичную тета-активность в лобно-центральной области, пароксизмальные проявления (пики или комплексы «пик-волна»), часто локализованные в задних отделах, вспышки медленных комплексов «пик-волна» (особенно во время сна) и прогрессирующее замедление и деградация фонового ритма (Niedermeyer et al., 1986, Glaze et al, 1987).
Частым проявлением является гипервентиляция с дыхательными паузами, иногда вызывающая синкопальные состояния, которые могут быть ошибочно приняты за эпилептические припадки. От двух третей до трех четвертей пациентов не способны к самостоятельному передвижению, а с прогрессированием четвертой стадии в большинстве случаев эта способность окончательно утрачивается (Hagberg, 1989). Патология роста встречается в большинстве случаев. Чрезвычайно часто встречающийся кифосколиоз является одним из основных осложнений синдрома. Устойчивый лабораторный маркер не выявлен. Первично зарегистрированная гипоаммониемия обнаруживается только в редких случаях и обычно обусловлена внешними факторами, например, лечением вальпроатами.
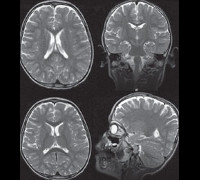
На МРТ выявляется уменьшение объема мозга, преимущественно за счет белого вещества, уменьшение объема хвостатого ядра и среднего мозга и нормальное строение извилин (Reiss et al., 1993).

г) Разновидности синдрома Ретта. Фенотип девочек с синдромом Ретта отличается большим разнообразием и варьирует от врожденных форм с практически полным отсутствием психического развития до легких форм, при которых может сохраняться способность к передвижению и даже речь (Huppke et al., 2003). Выделяют «скрытые формы», при которых проявления заболевания типичны, но выражены слабо; тяжелые врожденные формы, при которых практически полностью отсутствует психическое развитие; умеренные формы с регрессом в позднем детском возрасте; скрытые формы с широким диапазоном проявлений и варианты заболевания с сохранением речи (Hagberg и Skjeldal, 1994). В случае сохранения речи девочки могут обладать словарным запасом от 20 до нескольких сотен слов, а некоторые говорят длинными (обычно в виде эхолалий) предложениями (Zappella et al., 1998, 2001).
Также описана форма заболевания с проявлениями синдрома Ангельмана (Hitchins et al., 2004). Судорожный вариант заболевания характеризуется ранним началом припадков в виде инфантильных спазмов, которые могут предшествовать другим типам тонических или фокальных припадков и резистентны к лечению. Данная форма заболевания связана с мутациями гена CDKL5 (Weaving et al., 2004). Тем не менее, мутация данного гена выявляется не во всех случаях; вероятно, не все случаи мутации гена CDKL5 проявляются синдромом Ретта. Синдром Ретта был выявлен только у одного из семи пациентов с данной мутацией, у которых отмечались инфантильные спазмы и выраженная задержка умственного развития (Archer et al., 2006). Зарегистрированы редкие случаи типичного синдрома Ретта у мальчиков (Matsuyama et al., 2005). Среди мальчиков-носителей мутации чаще отмечается энцефалопатия новорожденных без типичных для синдрома проявлений (Moog et al., 2006).
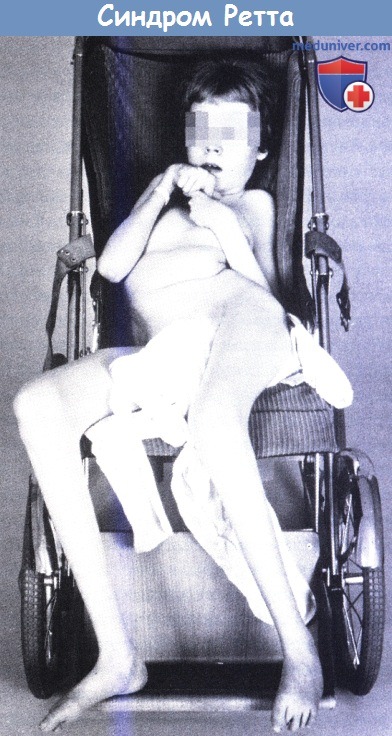
Синдром Ретта у 15-летней девочки.
Характерные стереотипные движения кистей, выраженный сколиоз и атрофия нижних конечностей.
д) Диагностика. Диагноз синдрома Ретта до сих пор устанавливается на основании анамнеза и клинических проявлений, а обнаружение мутации гена МЕСР2 во многих случаях служит подтверждением (Huppke и Gartner, 2005).
Диагностические критерии синдрома Ретта приведены в таблице ниже. У многих девочек с синдромом Ретта на ранних стадиях заболевания отмечаются аутистические проявления без четко выраженных неврологических отклонений. Поэтому в возрасте до трех лет часто ставится диагноз аутизма. Диагноз синдрома Ретта учитывается во всех случаях выявления симптомов аутизма у девочек в очень раннем детском возрасте. В исследовании Witt-Engerstrom и Gillberg (1987) девочек с синдромом Ретта в 80% случаев изначально предполагался аутизм или проявления аутизма, но на основе имеющихся данных о распространенности было установлено, что в 1/3-1/2 случаев выявления симптомов аутизма в течение первых лет жизни в итоге определялся симптом Ретта.
В редких случаях синдром Ретта характеризуется необычно медленным течением. В таких случаях диагноз «чистого аутизма» с серьезной/умеренной умственной отсталостью может не меняться в течение многих лет. Частая встречаемость атипичных форм (Goutieres и Aicardi 1985), отсутствие надежного маркера во многих случаях должны предостеречь от окончательного диагноза. Возможность выявления синдрома Ретта и задержки умственного развития у сибсов позволяет предположить наличие широкого спектра проявлений, но для подтверждения диагноза необходим определенный тест (Huppke et al., 2003). Hagberg и Skjeldal (1994) представили предварительные диагностические критерии для разновидностей синдрома Ретта.
е) Дифференциальная диагностика синдрома Ретта. В основном необходима дифференциальная диагностика с синдромом аутизма. Нейронный восковидный липофусциноз новорожденных с движениями рук, имитирующими вязание, описанный Santavuori, может иметь сходство с синдромом Ретта, но редко встречается за пределами Финляндии (Santavuori et al., 1973; Santavuori 1988). Дефицит орнитинтранскарбомилазы также ошибочно может приниматься за синдром Ретта. Синдром Ангельмана может иметь сходство с синдромом Ретта в связи с судорожной атаксией, наблюдаемой в обоих случаях помимо проявлений аутизма, задержки умственного развития и припадков. Исследование хромосом в сложных случаях позволяет дифференцировать заболевания.
Среди случаев с пограничными симптомами синдрома Ретта и аутизма (Gillberg, 1989), включая «скрытые формы» и формы заболевания с сохранением речи (Hagberg и Rasmussen, 1986), часто отмечаются многие классические проявления синдрома Ретта, но они не соответствуют всем критериям; обычно они также соответствуют большинству или всем критериям аутистического расстройства (или детского аутизма). У некоторых девочек классические симптомы аутизма проявляются только после длительного преморбидного периода (или 1 стадии) заболевания. Ретт-подобные симптомы встречаются также в сочетании с другими неврологическими расстройствами, такими как синдром Мебиуса (Gillberg и Steffenburg, 1989) и мукополисахаридоз.

ж) Лечение. Лечение синдрома Ретта неэффективно. При попытке применения бромокриптина и налоксона положительных результатов получено не было. Пациентам необходима физиотерапия и внимательное отношение к деталям обыденной жизни.
Стимуляторы показаны девочкам с хорошей реакцией на лечение в течение раннего периода отмены препарата. Важной частью терапии синдрома Ретта является ортопедическое лечение для предупреждения развития или уменьшения выраженности сколиоза.
Лечение поведенческих/психиатрических отклонений, вызванных синдромом Ретта, требует знания естественного течения заболевания, чтобы такие симптомы как аутизм и ночной смех не интерпретировались ошибочно как специфические психологические отклонения или проблемы общения. Восприятие речи при синдроме Ретта чрезвычайно снижено. Общение осуществляется с помощью зрительного контакта и жестов. Некоторые функции рук могут быть восстановлены при длительной ежедневной тренировке каждой руки в отдельности.
При применении бромокриптина (20 мг/кг в сутки) были достигнуты некоторые положительные результаты (Zappella et al., 1990), но для подтверждения эффективности необходимо проведение двойных слепых пла-цебо-контролируемых исследований. Результаты применения налоксона неоднозначны.
з) Исход. Отдаленные исходы синдрома Ретта известны только частично. Продолжительность жизни относительно увеличена, некоторые пациенты достигают возраста 80 лет и более. Подавляющее большинство (практически все) пациенты имеют чрезвычайно выраженные неврологические и/или умственные отклонения и зависят от других людей практически во всех областях повседневной жизни. В большинстве случаев клиническая картина осложняется эпилепсией, запорами, сколиозом, прогрессирующими двигательными (и вазомоторными) отклонениями. Психиатрические/по-веденческие отклонения могут являться предметом озабоченности в детском и иногда в подростковом возрасте, но обычно они в меньшей степени затрагивают пациентов старшей возрастной группы.
Синдром Ретта
Синдром Ретта – генетическое заболевание, характеризующееся нарушением развития нервной системы по причине отсутствия ингибирования определенных генов. Проявлениями этого состояния являются прогрессирующая умственная отсталость у девочек (при крайне редких атипичных формах – и у мальчиков), мышечная гипотония, атаксия, искривления позвоночника. Диагностика синдрома Ретта основывается на данных общего и неврологического осмотра, магнитно-резонансной томографии, электроэнцефалографии и молекулярно-генетических анализов. Специфического лечения не существует (имеются лишь определенные наработки с обнадеживающими результатами при опытах на животных), применяют симптоматическую терапию для облегчения состояния больного.
Общие сведения
Синдром Ретта – генетическое заболевание психоневрологического характера, практически всегда развивающееся у девочек и проявляющееся тяжелой степенью умственной отсталости. Эта патология была впервые выявлена еще в 1954 году австрийским неврологом А. Реттом, однако в качестве отдельной нозологической единицы он выделил данное заболевание лишь в 1966 году. Широкую известность в научном мире синдром Ретта получил в 1983 году после исследований Б. Хагберга. Это состояние является довольно распространенным – его встречаемость составляет примерно 1:10-15 тысяч новорожденных девочек, всего на сегодняшний день описано несколько десятков тысяч случаев патологии. Механизм наследования синдрома Ретта – доминантный, сцепленный с Х-хромосомой, именно поэтому он встречается практически всегда у девочек. У мальчиков из-за отсутствия парной Х-хромосомы генетические повреждения, приводящие к такому заболеванию, почти всегда являются летальными. Однако существует несколько атипичных форм синдрома Ретта, характеризующихся более сглаженной клинической картиной и поэтому поражающих лиц мужского пола. Кроме того, у мальчиков такая патология может развиться при наличии дополнительной Х-хромосомы – синдроме Клайнфельтера.
Причины синдрома Ретта
Этиология и патогенез синдрома Ретта достаточно сложны и обусловлены взаимодействием различных генов и их влиянием на развитие головного мозга человека. Первопричиной заболевания является нонсенс-мутация (по некоторым данным, к аналогичным нарушениям приводят и миссенс-мутации) гена MECP2, локализованного на Х-хромосоме, в результате чего его экспрессия полностью прекращается. Он кодирует специфический протеин под названием метил-CpG-связывающий белок 2, участвующий в регуляции транскрипции определенных участков ДНК. Данный белок содержит два домена, один из которых способствует его присоединению к метилированным участкам хромосом (которые расположены вблизи генов, регулирующих развитие головного мозга), а второй выступает как репрессор транскрипции. Причина синдрома Ретта как раз и заключается в отсутствии ингибирования некоторых генов, что приводит к нарушению формирования нервной ткани.
При этом синдром Ретта нельзя рассматривать как нейродегенеративное заболевание, так как при нем не наблюдается разрушения нейронов или нейроглии. Гистологические исследования тканей головного мозга больных выявляют нарушение ультраструктуры нервных клеток – уменьшение размеров, изменение количества дендритов, затрудненное образование нервных тканей. Объем нейроглии при синдроме Ретта снижен, в результате этого на макроскопическом уровне размер головного мозга тоже уменьшается на 20-30% по сравнению с возрастной нормой. Одной из причин вышеперечисленных процессов является отсутствие торможения выделения фермента GAD67 (ингибирование гена этого энзима осуществляется метил-CpG-связывающим белком 2), что, в свою очередь, приводит к увеличению концентрации тормозных трансмиттеров из группы ГАМК. В результате этого у больных синдромом Ретта наблюдается значительное превалирование процессов торможения в головном мозге, что отражается не только на физиологии центральной нервной системы, но и на ее морфологическом строении.
Врачами-генетиками было установлено, что полное отсутствие в геноме нормального гена MECP2 в подавляющем большинстве случаев является летальным состоянием и нередко приводит к внутриутробной смерти плода. Такое состояние имеет место у мальчиков (по причине наличия только одной Х-хромосомы) или у девочек-гомозигот, что встречается крайне редко. Из-за этого в половом распределении синдрома Ретта наблюдается абсолютное превалирование больных женского пола. Мутации гена MECP2 в большинстве случаев являются спонтанными или герминативными – предположительно, 70% случаев этого заболевания обусловлено генетическим дефектом Х-хромосомы в половых клетках отца. Дефекты этого гена приводят и другим патологиям центральной нервной системы – варианту Запела, синдрому Луба (Х-сцепленная умственная отсталость у мальчиков), врожденной энцефалопатии. Некоторые исследователи относят эти состояния к атипичным формам синдрома Ретта.
Симптомы синдрома Ретта
У новорожденных девочек синдром Ретта поначалу никак не проявляется, первые 6-12 месяцев развитие ребенка происходит обычными темпами без каких-либо отклонений. В дальнейшем прогрессирование заболевания характеризуется определенной стадийностью. Первая стадия синдрома Ретта, чаще всего возникающая в возрасте от 6-ти месяцев до 2,5 лет, характеризуется появлением у ребенка мышечной гипотонии, замедления психомоторного развития с последующим отставанием от сверстников, потерей интереса к играм и окружающим людям. Врачи-педиатры отмечают более медленный, нежели в норме, рост стоп и кистей в длину и замедление роста окружности головы. Иногда помимо неврологических проявлений может отмечаться нарушение работы печени, сердца, желудочно-кишечного тракта.
Вторая стадия синдрома Ретта характеризуется более выраженными клиническими проявлениями. Она развивается на протяжении 1-2 лет после появления первых симптомов заболевания, при этом у ребенка сначала наблюдается беспокойство, нарушения сна. Затем довольно быстро, всего за несколько недель, больные синдромом Ретта теряют практически все приобретенные до этого времени навыки – утрачивается речь, исчезает способность к ходьбе. Также для этой стадии развития патологии характерны расстройства дыхания – периоды апноэ по 1-2 минуты могут перемежаться с приступами учащенных и глубоких дыхательных движений (гипервентиляция). Дыхательные нарушения при синдроме Ретта отличаются наличием только при бодрствовании больного и отсутствием во время сна. Часто возникают многочисленные неврологические нарушения: атаксия, эпилептические припадки, часто повторяющиеся стереотипные движения.
Третья стадия синдрома Ретта называется псевдостационарной, так как при ней мало заметны признаки прогрессирования заболевания. Обычно она длится от 4 до 15 лет, состояние больных стабильно, однако наблюдаются судорожные приступы, глубокая умственная отсталость, гиперкинезы. В большинстве случаев синдрома Ретта этот этап оканчивается в пубертатном периоде. Четвертая стадия синдрома Ретта характеризуется уменьшением частоты эпилептических припадков вплоть до их исчезновения при прогрессировании двигательных расстройств. Большинство больных полностью теряют подвижность, возникает атрофия мышц, сосудистые нарушения в нижних конечностях, что может привести к развитию трофических язв. Из-за слабости мышечного корсета спины при синдроме Ретта возникает сколиоз или другие формы искривления позвоночника.
Диагностика
Диагностика синдрома Ретта производится на основании изучения анамнеза больного, его настоящего статуса, магнитно-резонансной томографии и энцефалографии, молекулярно-генетических анализов. Изучение наследственного анамнеза, как правило, не имеет особого смысла по причине спорадического характера мутаций гена MECP2. Характерными для синдрома Ретта являются нормальное развитие ребенка до 6-12 месяцев, возникновение мышечной гипотонии и беспокойства в раннем детстве, появление в дальнейшем атаксии и частых эпилептических припадков, стремительное утрачивание приобретенных навыков. В дальнейшем у больных регистрируется тяжелая умственная отсталость, мышечная слабость (вплоть до атрофии), искривление позвоночника, судорожные припадки.
При осмотре больных синдромом Ретта выявляется отставание в росте и его остановка, резкое уменьшение окружности головы, отсутствие речи (на начальных этапах патологии характерна эхолалия). На магнитно-резонансной томографии головного мозга обнаруживается уменьшение размера органа, нечеткая дифференциация серого и белого вещества, базальных ганглиев, снижение складчатости коры больших полушарий. Электроэнцефалограмма подтверждает снижение фоновой активности головного мозга и резко ослабленную реакцию на внешние раздражители.
Наиболее точную диагностическую информацию дают методы современной генетики – поиск делеций в локусе гена MECP2 или прямое секвенирование его последовательности для определения мутаций. Такое подтверждение синдрома Ретта возможно и в рамках пренатальной диагностики генетических заболеваний. Вспомогательную роль в установлении этого состояния может играть обследование внутренних органов (например, методами УЗИ) – у 20-30% больных выявляется недоразвитие печени или селезенки.
Лечение синдрома Ретта
Специфического лечения синдрома Ретта на сегодняшний день не существует. Имеются обнадеживающие данные некоторых исследовательских лабораторий, сотрудникам которых удалось «включить» ген MECP2 у мышей и тем самым добиться исчезновения симптомов заболевания. В сфере практической медицины пока доступна только симптоматическая терапия, однако и она сопряжена с рядом трудностей – в частности, эпилептические припадки при этом заболевании плохо поддаются устранению противосудорожными средствами. Также для лечения синдрома Ретта применяют ноотропные препараты, нарушения сна корректируют снотворными препаратами из группы барбитуратов или мелатонином.
Прогноз и профилактика
Прогноз синдрома Ретта неблагоприятный, так как это заболевание неуклонно ведет к тяжелой умственной отсталости, а также к ряду двигательных и неврологических нарушений. Пациенты с этой патологией при соответствующем уходе и симптоматическом лечении способны доживать до 40-50 лет, однако риск внезапной смерти у них довольно высокий. Значительно ухудшает прогноз синдрома Ретта и снижает продолжительность жизни больных наличие пороков развития внутренних органов, что имеет место примерно в трети случаев. Основная причина летального исхода – дыхательная или полиорганная недостаточность и внезапная смерть, у взрослых больных также велик риск инсульта. Профилактика синдрома Ретта возможна только в виде пренатальной диагностики этого заболевания генетическими методами. При наличии дефектной формы гена MECP2 у мальчика нарушение формирования головного мозга и внутренних органов можно заметить при профилактических ультразвуковых исследованиях во время беременности.
Синдром Ретта у детей: общие сведения и симптомы

Синдром Ретта – это генетическое заболевание, сопровождающееся тяжелыми психоневрологическими симптомами. Диагностика его затруднительна: оно практически никогда не обнаруживается внутриутробно, а после рождения проявляется не ранее, чем через 6 месяцев. Своему носителю оно грозит глубоким слабоумием, двигательными ограничениями, дезадаптацией в социуме.
Истоки заболевания
В масштабном формате о расстройстве заговорили в 1983 году благодаря шведскому ученому Бенгту Хагбергу. В это время он со своей группой изучал 35 подобных между собой случаев в 3 разных странах: в Португалии, Франции и Швеции.
Однако Хагберт не является первооткрывателем синдрома. Впервые его обнаружил педиатр Андреас Ретт, имя которого носит заболевание. Он наблюдал за двумя девочками, имеющими одинаковые симптомы. Их он заметил в очереди на прием. Они сидели на коленях у матерей, а те держали их за руки. Девочки раскачивались как маятники, а затем внезапно обе начали совершать стереотипные движения руками. Дети застыли в одном положении, отстраненные от окружающего мира. Взгляд был направлен в одну точку. Поражала их синхронность в движениях и поведении.
В своих письменных архивах врач отыскал подобные истории болезни, а затем отправился в Европу, чтобы разыскать и там таких же пациентов. В 1966 он сделал первые публикации своих исследований, которые, однако, не вызвали особого интереса.
Зафиксированную им болезнь Ретт назвал синдромом атрофии мозга. Сначала ее считали проявлением аутизма или шизофрении, и только лишь в 1983 году вывели в отдельную нозологическую единицу.

В настоящее время синдром относят к категории довольно редких генетических заболеваний. Он встречается с частотой случаев 1 на 15000. Причиной его называют мутацию гена МЕСР2. Этот ген отвечает за синтез определенного белка, влияющего на развитие мозга. В норме этот белок, спустя некоторое время после рождения, должен подавляться другими генами, чтобы обеспечить нормальное развитие мозга.
Если же ген МЕСР2 мутирован, то белок инактивируется не полностью, что вызывает аномальное мозговое созревание, и провоцирует развитие синдрома Ретта.
Обычно мутирующий ген располагается в Х хромосоме, потому заболеванием страдают преимущественно девочки.
Почему мальчики не болеют
Учитывая, что мутирующий ген несет в себе Х-хромосома, то девочки в плане заболевания находятся в более «выигрышной» позиции. У них присутствует две Х-хромосомы. Поэтому если одна из них «бракованная», то вторая функционирует нормально. Это дает девочке хоть малый шанс на нормальное существование.
У мальчика Х-хромосома одна. Если она имеет мутационный ген, значит, выпадает из работы полностью, и ее нечем заменить. Такие малыши мужского пола, как правило, погибают еще внутриутробно, так и не родившись. Поэтому синдром Ретта у мальчиков встречается крайне редко.

Но, несмотря на такую особенность заболевания, очень редко, но все-таки мальчики с подобным синдромом выживают. Это может быть связано с тем, что не все гены в Х-хромосоме подвергаются мутации. Из-за этого заболевание развивается не столь остро.
Другая причина – наличие у мальчика синдрома Клайнфельтера. При этом наблюдается полисомия половых хромосом, то есть их набор составляет ХХУ. И, если одна Х-хромосома имеет патологический ген, то вторая может регулировать синтез белка и дарить мальчику возможность жизни. Получается такая же картина, как и у девочки.
Как развивается заболевание
Синдром Ретта у детей – довольно коварное заболевание. При рождении оно практически не проявляет себя. Первые его симптомы появляются в период от 6 мес. до полутора лет. Однако некоторые, еле заметные признаки, в первом полугодии все-таки имеются. Но они настолько ничтожны, что не привлекают внимания.
Вот что говорит мама одной из девочек с синдромом по поводу первого полугодия ее жизни. Она придала значение этим мелочам только по прошествии 1 года и 7 месяцев с рождения ее дочери, когда проявления стали уже явными. Из предвестников болезни она отметила, что ее малышка начала держать голову в 3 месяца, а не в 2, как это положено. В 6 месяцев она еще не могла сидеть, а ходить начала только в 1 год и 4 месяца. Психологически развивалась нормально, и говорить начала рано, но это были не стандартные слова «мама», «папа», а «зайчик», «мишка» и др.
В 1 год и 7 мес. она перестала узнавать родителей и, казалось, не нуждалась в них. Весь день проводила за одним однообразным занятием: кидала мяч или катала коляску. Часами ходила по кругу, пока ее не останавливали или она запиналась. Такое стереотипное поведение носит название полевого, когда действие затягивает больного, и он не может ничего сделать.

В четыре года к симптомам присоединились эпилептоидные припадки. Однако по достижении школьного возраста девочка находилась на домашнем обучении, и делала некоторые успехи.
12–6 лет – это был период ремиссии, когда болезнь практически не беспокоила. Но с 16 лет появились новые, более глубокие проблемы, связанные с костными деформациями и болезнями внутренних органов. Одна нога девочки была короче другой почти на 10 см, что не могло не препятствовать ходьбе. В 20 лет она весила всего 24 кг с ростом 158 см.
Обычно СР протекает в 4 стадии.
Первая стадия, которая, как правило, стартует с 6 месяцев до полутора лет, проявляется нарастанием раздражительности и лабильностью настроения у ребенка. Эпизоды плача и психомоторного возбуждения сменяются все большей пассивностью. Малыш бесцельно передвигается по комнате, пропадает интерес к игрушкам. Но контакт с матерью сохраняется.
Вот как описывает женщина поведение своей дочери на заре заболевания: она кричала целый день без остановки, билась головой о стены, не могла уснуть. Что бы мы ни делали, она не успокаивалась. Это был настоящий ад. Но больше угнетало то, что ни один врач не мог поставить вразумительный диагноз.
Развивается диспропорция головы и конечностей по отношению к телу. Они становятся несоизмеримо маленькими. Замедляется рост, и снижается тонус мышц.
Вторая стадия, длящаяся несколько лет, отличается пестротой симптомов. Сразу обращает на себя внимание снижение интеллектуальных способностей, развивается умственное слабоумие. Происходит регресс практически всех полученных навыков. Речь полностью исчезает или переходит в степень эхолалии – механического повторения услышанного.
Приобретенные двигательные навыки, предметно-ролевое поведение теряются и замещаются двигательными стереотипами. Характерный симптом: многочисленно повторяющиеся движения, напоминающие мытье рук. Кроме этого, ребенок постоянно заламывает или потирает их, размахивает ими, хлопает в ладоши. Сжатие пальцев рук вполне нормально в 4 месяца, но в более позднем возрасте говорит об остановке развития. Малыш утрачивает хватательный рефлекс, не способен производить вращательные движения руками.
Постепенно двигательная активность сходит на нет. Нарушается походка, ребенок ходит, не сгибая коленей.

Третья стадия длится 10 лет и более, характеризуется она развитием стойкого, глубокого слабоумия, вплоть до идиотии. Наблюдается полная потеря способности говорить и понимать обращенную к ребенку речь. Появляется тремор всего тела, отягчающий движения. Усиливаются судорожные припадки.
Четвертая, конечная стадия – это период усугубления ранее проявляемых симптомов. Стойкая утрата умственных способностей, двигательных навыков, развитие мышечных дистрофий, приводящих к полному обездвиживанию.
Продолжительность жизни таких больных в среднем колеблется до 30 лет, хотя известны случаи, когда они доживали и до 50-летнего возраста.
Самые частые симптомы расстройства
Типичные симптомы для синдрома Ретта – мышечные и двигательные нарушения. Мышцы находятся в гипертонусе или же, наоборот, теряют его. В этом случае у ребенка развивается неправильное положение тела, прогрессирует частичные параличи и нарушение координации. Например, девочки скрещивают ноги во время ходьбы.
Синкинезии – патологические сокращения мышц, возникают вслед за произвольным движением: простая улыбка способна вызвать резкий взмах ногой. Такое явление постепенно приводит к повреждению суставов, сухожилий и связок, провоцирует ортопедические нарушения. Последние проявляются во всевозможных деформациях и также очень часто сопровождают таких детей. Среди них выделяют вывих тазобедренного сустава, провоцируемый малой подвижностью.
Статическая деформация стопы чаще развивается из-за нарушенного мышечного тонуса. Распространенной считается патология под названием «конская стопа», связанная со снижением подвижности голеностопного сустава. Ее можно узнать по пятке, которая не достигает земли, стопа при этом смещается кнаружи или вовнутрь. Причина патологии – гипертонус икроножной мышцы.
Сколиоз – боковое искривление позвоночника, который провоцирует массу проблем у таких пациентов: деформации суставов и костей, боли во время ходьбы, в стоячем или сидячем положении, утрата способности передвигаться. Сколиоз грудного отдела вызывает легочную недостаточность. Появляются также проблемы с пищеварением.
У детей с синдромом Ретта наблюдается повышенное слюнотечение. Но это происходит не из-за избытка количества слюны, а потери способности сглатывать ее.
Нарушение питания может развиваться из-за частых приступов тошноты. Она появляется на любые аспекты питания: на определенный продукт, его температуру, на способ приготовления. Так, ребенок способен отрицательно реагировать на пищу, поданную кусочками, или на комочки в блюде.

Постоянная тошнота провоцирует отказ от питания, а значит, потерю в весе.
Плохое сглатывание слюны, которая регулирует кислотность в желудке, и повышенное внутрибрюшное давление вызывают желудочно-пищеводный рефлюкс, то есть забрасывание содержимого желудка в пищевод. Это чревато такими последствиями, как воспаление стенки пищевода, респираторные инфекции.
Малоподвижный образ жизни, неврологические расстройства, неправильное питание провоцируют возникновение запоров у детей с синдромом Ретта. Они носят тяжелый характер, поскольку способны вызывать закупорку кишечника и сильные боли.
Повышенное слюнотечение, тошнота, рефлюкс снижают потребление ребенком пищи и даже развивают на нее негативную реакцию. В результате этого ребенок теряет в весе. Этот процесс стоит строго контролировать, поскольку он чреват истощением.
Другое тяжелое расстройство связано с работой дыхательной системы, развивающееся вплоть до приступов апноэ. Это явление настолько часто среди детей с синдромом, что нередко стает причиной их гибели.
Важными патогномоничными признаками синдрома считаются проявления аутизма. Именно из-за них заболевание изначально считали одной из форм этого расстройства, а в настоящее время относят к болезням аутистического спектра.
Аутистические признаки проявляются в отстранении от окружающего мира, в том числе и от родственников. Ребенок замыкается в себе, может не откликаться, когда его зовут. Предпочитает одиночество. Дети боятся чужих людей и непривычных ситуаций.
Лицо такого ребенка становится похожим на каменное. Взгляд блуждающий или устремлен в одну точку. Поведение часто непредсказуемо: случаются приступы неутомимого смеха или плача. Склонны к самоповреждениям: царапают кожу, кусают пальцы, вырывают волосы.
Нетипичная картина
Наряду с типичной формой заболевания, описанной выше, встречаются и атипичные формы. Они имеют свои особенности, от которых зависит тяжесть заболевания.
- Zapella – форма синдрома с неярко выраженными признаками. Речь частично сохранена, умеренно выражен сколиоз, умственная отсталость средней степени тяжести. Физически развиваются нормально.
- Hanefeld – в клинической картине преобладает раннее развитие судорожных приступов. Часто они случаются даже до появления умственной деградации.
- Rolando – на первый план выходят признаки задержки психомоторного развития. Ребенок теряет возможность передвигаться, нарастает стереотипия движений, его беспокоят дыхательные нарушения.

Синдром Ретта – сложное генетическое заболевание. Прежде всего, его сопровождает полная умственная деградация и психоневрологические нарушения, влекущие за собой многочисленные патологии других систем организма.
К сожалению, в мире еще не существует способа кардинального искоренения болезни, хотя ученые ведут постоянные разработки в этом направлении.
Лечение синдрома сводится к трем основным направлениям. Медикаментозная терапия назначается для купирования судорожных припадков и стимуляции работы головного мозга.
Диетотерапия включает в себя контроль массы тела, употребление в пищу высококалорийных, витаминизированных продуктов.
Однако наибольшее внимание уделяется реабилитационным мероприятиям, направленным на укрепление опорно-двигательного аппарата и поддержание умственного, психомоторного развития.
Важно сохранить комплексный, всесторонний подход к проблеме. Такие дети нуждаются в постоянной поддержке со стороны взрослых и веры в них. Сотрудничество с ними, как с полноценной ячейкой общества, способствует их лучшей адаптации в социуме и более благоприятному развитию.
Синдром Ретта
Синдром Ретта считается редким прогрессирующим генетическим заболеванием нервной системы. Синдром проявляется в раннем и быстром регрессе развития с последующим ухудшением ряда функций организма.
Болезнь носит имя врача – педиатра Андреаса Ретта, впервые описавшего проявления синдрома в 1966 году. На сегодняшний день его распространенность по разным данным составляет 1:10000 – 1: 15000 случаев.

Синдром Ретта – генетическое заболевание
Чтобы человеческий организм развивался, нужна информация: из чего будет построена клетка, как она будет действовать в случае изменения внешней среды, каким образом в конечном итоге положено работать тому или этому органу… Сведения о бесконечном множестве подобных “мелочей”, необходимых для правильного формирования невероятно сложного человеческого тела, содержатся в молекулах ДНК. Отдельные фрагменты ДНК называются генами. Они хранят зашифрованную информацию относительно того или иного признака организма. В свою очередь, гены “уложены” в хромосомы, набором которых укомплектована каждая наша клетка.
Отдельного внимания заслуживают половые хромосомы – они отличают женский пол от мужского, а также служат средством передачи информации о строении и функционировании организма от родителей к потомству. В норме женский организм содержит две Х-хромосомы, а мужской – Х и У. Из информации родительских Х и/или У хромосом, заключенных в половых клетках – сперматозоиде и яйцеклетке – зародыш складывает свой собственный генетический код, определяющий его дальнейшее развитие.
Поломка в какой-либо из родительских хромосом (Х или У) может привести к возникновению заболевания у ребенка, даже в случае, если сами родители внешне здоровы.
При синдроме Ретта, как правило, поломка находится в Х-хромосоме, и изменяет ген МеСР2, отвечающий за развитие нервной системы, в том числе головного мозга, что ведет к проявлению болезни у девочек. У мальчиков в случае такого нарушения в единственной Х-хромосоме, не имеющей здорового дублера, болезнь оказывается летальной, такие дети гибнут вскоре после рождения. В редких случаях в мужском организме может иметься “лишняя” Х-хромосома, что позволит избежать гибели и проявиться заболеванию. Крайне редко у детей могут встречаться атипичные формы синдрома Ретта с более “мягкими” проявлениями.
Синдром Ретта у детей. Как заметить?
В большинстве случаев до 6 – 18 месяцев болезнь никак себя не обнаруживает. В раннем возрасте родители порой обращают внимание на вялость ребенка, потливость, слабый мышечный тонус, что относится к неспецифической симптоматике, то есть может наблюдаться при множестве разных состояний.
Как уже было сказано, суть синдрома Ретта – поражение нервной системы. “Сломанный” ген отвечает за формирование связи нервных клеток друг с другом (синаптогенез), а потому и внешние проявления синдрома будут выражены прежде всего в нарушении психоневрологического развития.
Стадии болезни
В своем развитии заболевание проходит 4 последовательные стадии.
I стадия (аутистическая) проявляется после 6-18 месяцев, и сопровождается отставанием в формировании возрастных навыков. К примеру, малыш не сидит, не встает или не ползает, не начинает говорить. Ранее приобретенные навыки (указательный жест, интерес к игрушкам и пр.) пропадают. Появляется “отрешенность” от внешнего мира, “уход в себя”, снижается зрительный контакт, часто замедляется рост головы.
Регулярное наблюдение у врача-педиатра, который отслеживая динамику развития ребенка, нередко первым обращает внимание на несоответствие возрастным показателям и может рекомендовать родителям более полное, подробное обследование у специалистов узкого профиля.
Первая стадия болезни длится от нескольких месяцев до года, затем сменяется так называемой “быстрой деструкцией” или “быстрым регрессом”.
II стадия (“быстрая деструкция”) начинается в 1-4 года и длится от нескольких недель до нескольких месяцев. По мере прогрессирования поражения нервной системы появляется все больше симптомов:
● усиление аутистической отрешенности, снижение интереса к общению;
● трудности поведения: импульсивность, неусидчивость, суетливость:
● утрата ранее приобретенных навыков разговорной речи;
● двигательные расстройства: утрата произвольных (управляемых сознанием) движений рук (захват и удержание предмета и пр.), смена ведущей руки, присоединение стереотипных движений руками (заламывание, хлопанье, постукивание, скручивание, потирание, “моющие” движения, поднесение руки ко рту, битье по подбородку, заведение рук назад).
Все это – свидетельства распада двигательных актов, регулируемых корой головного мозга.
В дальнейшем к вышеописанному присоединяются признаки нарушения работы мозжечка – атаксия походки (расстройство координации движений, ребенок становятся неловкими, нарушается равновесие, при ходьбе дети широко расставляют ноги и шатаются); тремор (мышечные сокращения, “дрожание”).
Утрачивается способность менять позу, вставать и садиться.
● нарушение дыхания (во время бодрствования остановка дыхания сменяется интенсивным, учащенным дыханием, зачастую сопровождаемым криком, может наблюдаться форсированное (усиленное) изгнание воздуха и слюны).
● расстройства настроения: беспричинные перепады от веселости до тоски, страхи и тревога, спонтанный плач, раздражительность.
● периферические вазомоторные расстройства (синюшность кистей и стоп, их зябкость и холодность на ощупь).
Наряду с этими признаками во II стадии синдрома Ретта может быть: скрежет зубов, расстройства сна, снижение реакции на боль, задержка роста.
III стадия (псевдостационарная) начинается с 2-10 лет и длиться годами. В это время наблюдается некоторое смягчение определенных симптомов: уходит аутистическая отрешенность, частично восстанавливается способность к общению, появляется понимание речи и жестов, в собственной речи – возникают отдельные слова.
Сглаживаются поведенческие сложности, улучшается общий фон настроения, несколько повышаются показатели внимания.
Параллельно формируются серьезные ортопедические нарушения (деформируются конечности, у ребенка появляется вывих тазобедренного сустава; вследствие прогрессирования неврологической симптоматики и атрофии мышц спины изменяется осанка, что ведет к усугублению расстройств дыхания и пищеварения).
Могут возникать эпилептиформные приступы: от так называемых “абсансов” с кратковременной утратой сознания и остановкой взора, до “больших припадков” с выраженными судорожными проявлениями.
IV (тотальная деменция) длится до десятков лет. Нарастает мышечная скованность, снижается подвижность вплоть до полной утраты навыков ходьбы.
Усугубляющиеся расстройства глотания и жевания могут приводить к отказу от твердой пищи, избирательности рациона, что служит причиной изменения веса (как снижения, так и увеличения), дефицита витаминов и минералов, утяжеления состояния ребенка.
Упомянутые расстройства глотания являются причиной слюнотечения, что в свою очередь способно опосредованно провоцировать гастроэзофагальный рефлюкс (забрасывание содержимого из желудка в пищевод) и запускать последующие нарушения работы желудочно-кишечного тракта.
Интеллектуальное развитие детей с синдромом Ретта на протяжении жизни остается на уровне умственной отсталости, а это значит, что такие дети будут постоянно нуждаться в уходе и заботе со стороны окружающих, а также специальном психолого-педагогическом сопровождении.
Атипичные формы синдрома Ретта
При атипичных формах синдрома Ретта IV стадии болезни обычно не наступает, и способность к ходьбе не утрачивается. Отрешенность от внешнего мира может сохраняться в течение всей жизни. В речи этих детей могут оставаться слова и короткие фразы в виде повторов – эхолалий и эхофразий. Возможно раннее проявление судорожного синдрома.
Постановка диагноза и лечение синдрома Ретта
Ставится исключительно врачами после полного обследования на основании сведений анамнеза, клинических признаков, данных лабораторно-инструментальных исследований, включающих ДНК-тестирование. Некоторые проявления синдрома Ретта схожи с признаками ряда других болезней, потому в вопросе диагностики важно комплексное обследование с консультацией таких врачей, как психиатр, невролог, генетик.
Дифференциальный диагноз проводится с аутизмом, детским церебральным параличом, синдромом Ангельмана, болезнями обмена веществ, умственной отсталостью.
К моменту написания этого материала специфического лечения синдрома Ретта не существует. С целью абилитации активно применяются психокоррекционные занятия, работа с дефектологами и логопедами, семейная психотерапия. При необходимости под строгим врачебным контролем используются медикаментозные средства, направленные на коррекцию тяжелых поведенческих трудностей, судорожных припадков, дыхательных расстройств, нарушений настроения и сна.
Учитывая, что синдрому Ретта часто сопутствует соматическая патология, то есть нарушение работы внутренних органов, важно помнить о необходимости дальнейшего наблюдения не только у психиатра, невролога и генетика, но еще и педиатра, ортопеда, гастроэнтеролога.
Римма Кондратьева, врач-психиатр Адаптационного отделения Центра им. Г.Е. Сухаревой ДЗМ
Синдром Ретта

Синдром Ретта – прогрессирующее дегенеративное заболевание центральной нервной системы, названное по имени ученого, который впервые описал эту патологию.
Диагностируется синдром в течение первых 2-х лет жизни ребенка, причем возникает он обычно у детей при нормально протекающей беременности, родах и полноценном развитии в первые месяцы жизни (иногда до 1,5 лет). Затем происходит остановка развития и регресс всех форм психической деятельности, что сопровождается возникновением аутизма, моторной стереотипии, прогрессирующего моторного снижения. В последующем это заболевание приводит к инвалидности и даже к смерти.
Основными проявлениями болезни являются обратное развитие уже приобретенных двигательных и речевых навыков в возрасте от 1,5 до 3-4 лет, повторяющиеся стереотипные и неконтролируемые движения рук, умственная отсталость.

Что это такое?
Синдром Ретта — психоневрологическое наследственное заболевание, встречается почти исключительно у девочек с частотой 1:10000 — 1:15000, является причиной тяжёлой умственной отсталости у девочек.
Впервые болезнь была описана австрийским неврологом Андреасом Реттом (нем. Andreas Rett) в 1966 году. Развитие ребёнка до 6 — 18 месяцев протекает нормально, но потом у девочки начинают пропадать приобретённые речевые, двигательные и предметно-ролевые навыки.
Характерным для данного состояния являются стереотипные, однообразные движения рук, их потирание, заламывание, при этом не носящие целенаправленного характера. Речь затрудняется, ответы становятся однообразными или эхолалическими, временами речь совсем пропадает (мутизм).
Причины возникновения
В 90-х годах существовала гипотеза о том, что синдром Ретта – это определённое нарушение, которое связано с генными мутациями, локализующимися в Х-хромосоме; обусловленное доминантным признаком и у мальчиков не может совмещаться с жизнью. В дальнейшем подтвердилась передача гена-мутанта Х-хромосомой отца тем, что это наследственная патология очень редко может встретиться у мальчиков, так как они получают от отца Y-хромосому. Именно поэтому при семейном типе наследования синдрома Ретта, мальчики в таких семьях рождаются практически здоровыми.
В настоящее время существуют подтверждения о наследственной природе заболевания. Генетическую причину возникновения синдрома Ретта связывают с изменённой Х-хромосомой и мутациями, которые происходят в генах, регулирующих процесс репликации. В данном случае происходит дефицит некоторых белков, которые регулируют этот рост, а также нарушается холинергическая их функция.
Для синдрома Ретта была выдвинута гипотеза о прерванном развитии, для которого характерен дефицит нейротрофических факторов. Таким образом, поражаются базальные ганглии, нижние моторные нейроны, вовлекается спинной мозг и гипоталамус. Анализируя морфологические изменения, учёные пришли к выводу, что происходят замедления в развитии мозга с самого рождения, который полностью останавливается в росте к четырём годам. А также у таких детей отмечается замедление в росте тела и некоторых соматических органов.
Стадии развития
Синдром Ретта формируется в течение длительного времени, поэтому заболевание принято подразделять на несколько стадий в зависимости от ухудшения состояния больного:
- Стагнация — временное приостановление болезни, при которой не происходит нарастания симптомов. Длится от 6-18 месяцев и более. Ребенок теряет интерес к окружающим событиям, заметна гипотония мышц, замедление роста головы и конечностей.
- Ухудшение состояния. Стадия длится от 1 года до 3-4 лет. Если ребенок овладел навыками речи, передвижения, они постепенно исчезают. Появляются стереотипные манипуляции руками, нарушения со стороны легочной системы (гипервентилляция, одышка, внезапная остановка дыхания), дискоординация движений, немотивированное беспокойство. Уже на 2 стадии появляются судорожные припадки, лечение которых не является результативным.
- Относительная стабильность, эта стадия может продолжаться вплоть до раннего школьного возраста. Отмечается умственная недостаточность, судороги, малая прибавка веса, нарушение эмоционального контакта с окружающими. Эпилептические припадки сменяются заторможенностью нервной системы.
- Завершающая стадия характеризуется снижением частоты судорог, зато появляется кахексия, сколиоз, выраженные нарушения со стороны дыхания. Возможна несостоятельность передвижения, определяют низкий рост конечностей и малую окружность головы.
Характерные симптомы, присущие той или иной стадии, не являются на сто процентов условными и могут варьироваться в зависимости от скоротечности болезни и индивидуальных клинических случаев.

Симптомы синдрома Ретта
Отдельно следует акцентировать внимание на основных симптомах, по которым определяется синдром Ретта, поскольку в медицинской практике были известны случаи, когда из-за неверной трактовки признаков болезни ставился совершенно другой диагноз, что приводило в итоге к быстрому летальному исходу.
Синдром Ретта определяется по таким критериям:
- Ярко выраженная микроцефалия. В период после рождения у ребенка наблюдается нормальное соотношение размера головы с туловищем. Постепенно рост головы замедляется, что вызвано уменьшением размеров головного мозга.
- Развивающийся сколиоз. Нарушения позвоночного отдела характерны для всех детей, страдающих данным недугом. Причиной искривления спины является дистония мышц.
- Ментальное развитие. Синдром Ретта характеризуется глубокой умственной отсталостью и отсутствием насыщенной познавательной деятельности, которая обычно присутствует у всех маленьких детей. Многие пациенты поначалу приобретают навыки говорения и восприятия окружающего мира, однако со временем полностью их теряют. У ребенка заметно отсутствие любого экспрессивного или импрессивного общения с окружением. Для определения нарушений в ментальном развитии специалисты используют стандартизированные психологические тесты.
- Специфические движения рук. У детей пропадают навыки держания каких-либо предметов, будь то игрушка или бутылочка с молоком. При этом возникают монотонные движения рук, напоминающие мытье под краном, а также характеризующиеся сжиманием, перебиранием пальцев, хлопками на уровне груди, лица и за спиной. Также пациент может сосать или покусывать руки, хаотично ударять себя ими по разным частям тела.
- Судорожные припадки. Практически в 80% случаев девочки страдают от эпилептических припадков, которые также сопровождаются парциальными приступами, дроп-атаками, тонико-клоническим типом припадка. Синдром Ретта характеризуется такими признаками формирования предприпадочного состояния: тремор, затрудненность дыхания, резкие движения, направленность взгляда в одну точку с полным при этом онемением тела. Данные симптомы нередко лечатся антиконвульсантами, однако подобные препараты не дают никакого положительного эффекта, поскольку перечисленные признаки не относятся к группе судорожных заболеваний, а представляют собой лишь синдром Ретта.
- Нейрохимические симптомы. Во время исследования пациентов, скончавшихся на тяжелой стадии синдрома Ретта, ученые определили, что размеры их мозга были меньше нормы на 12% или 24%, в зависимости от возрастной группы, в которой находился пациент. В мозжечке, коре полушарий головного мозга и спинальных ганглиях наблюдался дефицит нейронов и глиоз, а также пониженный уровень пигментации. Согласно морфологическим исследованиям к четырем годам у людей, страдающих синдромом Ретта, диагностировалась полная остановка развития мозга и замедление роста других органов и частей тела.
Диагностика
Консультация психиатра начинается со сбора анамнеза. Во время беседы с родителями специалист выясняет:
- нормально ли протекали беременность и роды;
- как развивался ребёнок на протяжении первых 6–18 месяцев;
- какова динамика роста головы;
- когда ребёнок начал терять приобретённые навыки;
- имеются ли у пациента стереотипные движения рук, судорожные припадки, нарушения речи, дыхания, походки и координации, задержка психомоторного развития.
Для точной диагностики врач часто назначает дополнительные обследования:
- ЭЭГ (электроэнцефалограмма), измеряющая биоэлектрическую активность головного мозга (медленный фоновый ритм — свидетельство мутации в Х-хромосоме);
- КТ головного мозга, позволяющее обнаружить изменения, указывающие на прекращение развития мозга;
- УЗИ внутренних органов, дающее понятие о степени их развития.
При отсутствии данных обследований синдром Ретта можно спутать с аутизмом. Для обоих этих состояний характерны:
- снижение способности к обучению;
- потеря речи;
- отсутствие контроля за тазовыми органами;
- уход от внешнего мира;
- отсутствие зрительного контакта;
- нежелание идти на эмоциональный и социальный контакт;
- нарушение чувствительности тела;
- беспричинные крики и плач.
Отличить одно заболевание от другого можно с помощью дифференциальной диагностики, разработанной в 1988 году на Международной конференции по синдрому Ретта.
Данные дифференциальной медицинской диагностики:
| Описание симптома | Проявление при синдроме Ретта | Проявление при раннем аутизме |
| Замедленный рост кистей рук, стоп и головы | Характерный признак синдрома Ретта | Признак отсутствует |
| Отставание в развитии в возрасте от шести месяцев до года | Не проявляется | Часто наблюдается |
| Дыхательные расстройства | Часто наблюдаются | Не проявляются |
| Стереотипные движения рук | Повторяющиеся однообразные движения рук в области пояса | Разнообразные и сложные движения, не ограниченные зоной пояса |
| Эпилептические припадки | Часто повторяются | Редко проявляются |
| Координация движений | Прогрессирует нарушение координации, переходящее в полную неподвижность | Движения и походка почти нормальные, но кажутся манерными |
Используя дифференциальную диагностику, отличить аутизм от синдрома Ретта могут не только врачи, но и сами родители.
Лечение
На данном этапе развития медицины синдром Ретта является неизлечимым заболеванием. Однако с помощью лекарственных препаратов, реабилитационных методик и специальной диеты можно добиться улучшения состояния ребёнка, предупредить серьёзные деформации тела и улучшить качество жизни больного.
Для улучшения общего состояния и смягчения симптомов врач может прописать следующие медикаменты:
- противопаркинсонические препараты (Бромокриптин, Перлодел);
- ноотропы для улучшения работы мозга (Кортексин, Церебролизин, Цераксон);
- лекарства для регулирования биологического режима дня и ночи (Мелатонин);
- препараты для успокоения нервной системы и коррекции поведения (Ноофен, Фенибут, Глицин);
- противоэпилептические препараты для уменьшения количества приступов (Карбамазепин, Ламотриджин);
- средства для поддержки работы внутренних органов: сердца, печени, желудка, кишечника, поджелудочной железы.
При сильно выраженной эпилепсии приём антиконвульсантов может оказаться неэффективным. Часто дети с синдромом Ретта сами «перерастают» приступы: к 10 годам припадки становятся редкими, а иногда и вовсе уходят.

Реабилитация
В программу реабилитации может входить:
- Консультации психолога и логопеда — один раз в неделю.
- Музыкотерапия — повышает коммуникативную активность ребёнка.
- Массаж и лечебно-оздоровительная физкультура — направлены на укрепление мышц, увеличение их тонуса.
Хорошие отзывы имеет метод, придуманный французским отоларингологом Альфредом Томатисом. Целью является повторное обучение ребёнка процессу слушания, что улучшает способности к изучению и освоению языков, увеличивает творческий потенциал и положительно влияет на социальное поведение малыша.
- Арт-терапия и дельфинотерапия — положительно воздействуют на психоэмоциональное состояние ребёнка.
- Иппотерапия (лечебная верховая езда) и гидротерапия (душ, обливания, обтирания) — оказывают биомеханическое воздействие на тело ребёнка, укрепляет мышцы.
- АВА-терапия — улучшает социальную адаптацию больных. Какие-то сложные действия (контактность, творческая игра, речь) для ребёнка разбиваются на мелкие части. В будущем они соединяются в один большой блок.
Стоит отметить, что хорошее терапевтическое действие оказывает посещение остеопата. Улучшение состояния наблюдается уже после нескольких сеансов.
Особенности питания
Питание больного ребёнка представляет определённые сложности. У многих девочек наблюдается повышенное слюнотечение, плохое состояние ротовой полости, поэтому накормить их настоящая проблема. Некоторые дети имеют хороший аппетит и с удовольствием употребляют любимые продукты. Но все они едят очень медленно, процесс приёма пищи может растягиваться до полутора часов. Что касается питья, то почти у половины больных малышей есть трудности с глотанием, которые проявляются поперхиванием, кашлем и могут грозить попаданием жидкости в дыхательные пути.
- Детям с синдромом Ретта тяжело пережёвывать пищу, содержащую грубые волокна (мясо, сырые овощи, фрукты), поэтому её нужно измельчать и давать в виде пюре. Гарниры лучше предлагать мелкими кусочками или в размятом виде. Во время кормления нужно следить, чтобы голова ребёнка была под правильным углом, не запрокидывалась.
- В некоторых случаях, когда процесс поглощения пищи становится слишком проблематичным и болезненным, имеет смысл кормить ребёнка через зонд специальными питательными смесями. Такой вариант позволяет значительно улучшить качество жизни малыша и может стать настоящим для него спасением.
Многие дети страдают от тошноты и часто отказываются от еды, поэтому накормить их бывает очень трудно, такие малыши быстро теряют в весе. Поэтому пища должна быть обогащённой белками и жирами, достаточно калорийной и витаминизированной. Рекомендуется кормить ребёнка малыми порциями и часто (каждые 3 часа), чтобы не перегружать пищеварительную систему. Детей младшего возраста поят витаминизированным молоком или молочными смесями.
Прогноз
Чтобы найти адекватное лечение, медики и ученые по всему миру ведут интенсивные исследования детского заболевания, называемого «синдром Ретта». Данные и результаты научных центров, занимающихся этой проблемой, уже подтверждают версию о том, что процессы, которые запускаются патологией, обратимы.
Ведется активная разработка стратегии использования стволовых клеток, на которых будет основано лечение синдрома Ретта. Уже сегодня предварительные средства опробованы на лабораторных мышах. Опыты профессора Беличенко, которые проводились в Калифорнийском университете, дают положительный прогноз и надежду на скорое научное открытие эффективного лечения синдрома Ретта.
Профилактика
Лечебная физкультура — один из оптимальных способов коррекции двигательных расстройств. Она включает упражнения, направленные на поддержание гибкости и амплитуды движений конечностей, а также как можно более длительное сохранение навыка ходьбы.
Предлагаются психологические программы максимального развития оставшихся сохранными двигательных навыков и формирования на их основе “языка общения”. Используется также музыкальная терапия, так как она оказывает благоприятный успокаивающий эффект на детей и частично компенсирует нарушение контакта с окружающим миром. Исследования синдрома Ретта интенсивно ведутся во всем мире, и открытие его специфического биологического маркера является, вероятно, лишь вопросом времени.
Когда это произойдет, возникнут новые перспективы лечения патологии или облегчения состояния больных, а также возможности пренатального скрининга и предотвращения этого тяжелого заболевания.
Синдром Ретта у девочек, детей. Что это такое, симптомы, описание, лечение, прогноз, причины
Ожидайте
Специалист свяжется с Вами сразу в рабочее время с
Пн – Пт с 10:00 – 19:00 МСК
Перезвоните мне

Ваш персональный менеджер: Екатерина
Ответственная и отзывчивая! 😊
Ожидайте
Специалист свяжется с Вами сразу в рабочее время, ежедневно с 10:00 – 19:00 МСК
Перезвоните мне

Синдром Ретта – это генетическое заболевание, сопровождающееся тяжелыми психоневрологическими симптомами.
Бесплатные занятия с логопедом
Бесплатный курс ИКТ для детей
Синдром Ретта – это генетическое заболевание, сопровождающееся тяжелыми психоневрологическими симптомами. Диагностика его затруднительна: оно практически никогда не обнаруживается внутриутробно, а после рождения проявляется не ранее, чем через 6 месяцев. Своему носителю оно грозит глубоким слабоумием, двигательными ограничениями, дезадаптацией в социуме.
ИСТОКИ ЗАБОЛЕВАНИЯ
В масштабном формате о расстройстве заговорили в 1983 году благодаря шведскому ученому Бенгту Хагбергу. В это время он со своей группой изучал 35 подобных между собой случаев в 3 разных странах: в Португалии, Франции и Швеции.
Однако Хагберт не является первооткрывателем синдрома. Впервые его обнаружил педиатр Андреас Ретт, имя которого носит заболевание. Он наблюдал за двумя девочками, имеющими одинаковые симптомы. Их он заметил в очереди на прием. Они сидели на коленях у матерей, а те держали их за руки. Девочки раскачивались как маятники, а затем внезапно обе начали совершать стереотипные движения руками. Дети застыли в одном положении, отстраненные от окружающего мира. Взгляд был направлен в одну точку. Поражала их синхронность в движениях и поведении.
В своих письменных архивах врач отыскал подобные истории болезни, а затем отправился в Европу, чтобы разыскать и там таких же пациентов. В 1966 он сделал первые публикации своих исследований, которые, однако, не вызвали особого интереса.
Зафиксированную им болезнь Ретт назвал синдромом атрофии мозга. Сначала ее считали проявлением аутизма или шизофрении, и только лишь в 1983 году вывели в отдельную нозологическую единицу.
В настоящее время синдром относят к категории довольно редких генетических заболеваний. Он встречается с частотой случаев 1 на 15000. Причиной его называют мутацию гена МЕСР2. Этот ген отвечает за синтез определенного белка, влияющего на развитие мозга. В норме этот белок, спустя некоторое время после рождения, должен подавляться другими генами, чтобы обеспечить нормальное развитие мозга.
Если же ген МЕСР2 мутирован, то белок инактивируется не полностью, что вызывает аномальное мозговое созревание, и провоцирует развитие синдрома Ретта.
Обычно мутирующий ген располагается в Х хромосоме, потому заболеванием страдают преимущественно девочки.
ПОЧЕМУ МАЛЬЧИКИ НЕ БОЛЕЮТ
Учитывая, что мутирующий ген несет в себе Х-хромосома, то девочки в плане заболевания находятся в более «выигрышной» позиции. У них присутствует две Х-хромосомы. Поэтому если одна из них «бракованная», то вторая функционирует нормально. Это дает девочке хоть малый шанс на нормальное существование.
У мальчика Х-хромосома одна. Если она имеет мутационный ген, значит, выпадает из работы полностью, и ее нечем заменить. Такие малыши мужского пола, как правило, погибают еще внутриутробно, так и не родившись. Поэтому синдром Ретта у мальчиков встречается крайне редко.
Но, несмотря на такую особенность заболевания, очень редко, но все-таки мальчики с подобным синдромом выживают. Это может быть связано с тем, что не все гены в Х-хромосоме подвергаются мутации. Из-за этого заболевание развивается не столь остро.
Другая причина – наличие у мальчика синдрома Клайнфельтера. При этом наблюдается полисомия половых хромосом, то есть их набор составляет ХХУ. И, если одна Х-хромосома имеет патологический ген, то вторая может регулировать синтез белка и дарить мальчику возможность жизни. Получается такая же картина, как и у девочки.
КАК РАЗВИВАЕТСЯ ЗАБОЛЕВАНИЕ
Синдром Ретта у детей – довольно коварное заболевание. При рождении оно практически не проявляет себя. Первые его симптомы появляются в период от 6 мес. до полутора лет. Однако некоторые, еле заметные признаки, в первом полугодии все-таки имеются. Но они настолько ничтожны, что не привлекают внимания.
Вот что говорит мама одной из девочек с синдромом по поводу первого полугодия ее жизни. Она придала значение этим мелочам только по прошествии 1 года и 7 месяцев с рождения ее дочери, когда проявления стали уже явными. Из предвестников болезни она отметила, что ее малышка начала держать голову в 3 месяца, а не в 2, как это положено. В 6 месяцев она еще не могла сидеть, а ходить начала только в 1 год и 4 месяца. Психологически развивалась нормально, и говорить начала рано, но это были не стандартные слова «мама», «папа», а «зайчик», «мишка» и др.
В 1 год и 7 мес. она перестала узнавать родителей и, казалось, не нуждалась в них. Весь день проводила за одним однообразным занятием: кидала мяч или катала коляску. Часами ходила по кругу, пока ее не останавливали или она запиналась. Такое стереотипное поведение носит название полевого, когда действие затягивает больного, и он не может ничего сделать.
В четыре года к симптомам присоединились эпилептоидные припадки. Однако по достижении школьного возраста девочка находилась на домашнем обучении, и делала некоторые успехи.
12–6 лет – это был период ремиссии, когда болезнь практически не беспокоила. Но с 16 лет появились новые, более глубокие проблемы, связанные с костными деформациями и болезнями внутренних органов. Одна нога девочки была короче другой почти на 10 см, что не могло не препятствовать ходьбе. В 20 лет она весила всего 24 кг с ростом 158 см.
Обычно СР протекает в 4 стадии.
Первая стадия, которая, как правило, стартует с 6 месяцев до полутора лет, проявляется нарастанием раздражительности и лабильностью настроения у ребенка. Эпизоды плача и психомоторного возбуждения сменяются все большей пассивностью. Малыш бесцельно передвигается по комнате, пропадает интерес к игрушкам. Но контакт с матерью сохраняется.
Вот как описывает женщина поведение своей дочери на заре заболевания: она кричала целый день без остановки, билась головой о стены, не могла уснуть. Что бы мы ни делали, она не успокаивалась. Это был настоящий ад. Но больше угнетало то, что ни один врач не мог поставить вразумительный диагноз.
Развивается диспропорция головы и конечностей по отношению к телу. Они становятся несоизмеримо маленькими. Замедляется рост, и снижается тонус мышц.
Вторая стадия, длящаяся несколько лет, отличается пестротой симптомов. Сразу обращает на себя внимание снижение интеллектуальных способностей, развивается умственное слабоумие. Происходит регресс практически всех полученных навыков. Речь полностью исчезает или переходит в степень эхолалии – механического повторения услышанного.
Приобретенные двигательные навыки, предметно-ролевое поведение теряются и замещаются двигательными стереотипами. Характерный симптом: многочисленно повторяющиеся движения, напоминающие мытье рук. Кроме этого, ребенок постоянно заламывает или потирает их, размахивает ими, хлопает в ладоши. Сжатие пальцев рук вполне нормально в 4 месяца, но в более позднем возрасте говорит об остановке развития. Малыш утрачивает хватательный рефлекс, не способен производить вращательные движения руками.
Постепенно двигательная активность сходит на нет. Нарушается походка, ребенок ходит, не сгибая коленей.
Третья стадия длится 10 лет и более, характеризуется она развитием стойкого, глубокого слабоумия, вплоть до идиотии. Наблюдается полная потеря способности говорить и понимать обращенную к ребенку речь. Появляется тремор всего тела, отягчающий движения. Усиливаются судорожные припадки.
Четвертая, конечная стадия – это период усугубления ранее проявляемых симптомов. Стойкая утрата умственных способностей, двигательных навыков, развитие мышечных дистрофий, приводящих к полному обездвиживанию.
Продолжительность жизни таких больных в среднем колеблется до 30 лет, хотя известны случаи, когда они доживали и до 50-летнего возраста.
САМЫЕ ЧАСТЫЕ СИМПТОМЫ РАССТРОЙСТВА
Типичные симптомы для синдрома Ретта – мышечные и двигательные нарушения. Мышцы находятся в гипертонусе или же, наоборот, теряют его. В этом случае у ребенка развивается неправильное положение тела, прогрессирует частичные параличи и нарушение координации. Например, девочки скрещивают ноги во время ходьбы.
Синкинезии – патологические сокращения мышц, возникают вслед за произвольным движением: простая улыбка способна вызвать резкий взмах ногой. Такое явление постепенно приводит к повреждению суставов, сухожилий и связок, провоцирует ортопедические нарушения. Последние проявляются во всевозможных деформациях и также очень часто сопровождают таких детей. Среди них выделяют вывих тазобедренного сустава, провоцируемый малой подвижностью.
Статическая деформация стопы чаще развивается из-за нарушенного мышечного тонуса. Распространенной считается патология под названием «конская стопа», связанная со снижением подвижности голеностопного сустава. Ее можно узнать по пятке, которая не достигает земли, стопа при этом смещается кнаружи или вовнутрь. Причина патологии – гипертонус икроножной мышцы.
Сколиоз – боковое искривление позвоночника, который провоцирует массу проблем у таких пациентов: деформации суставов и костей, боли во время ходьбы, в стоячем или сидячем положении, утрата способности передвигаться. Сколиоз грудного отдела вызывает легочную недостаточность. Появляются также проблемы с пищеварением.
У детей с синдромом Ретта наблюдается повышенное слюнотечение. Но это происходит не из-за избытка количества слюны, а потери способности сглатывать ее.
Нарушение питания может развиваться из-за частых приступов тошноты. Она появляется на любые аспекты питания: на определенный продукт, его температуру, на способ приготовления. Так, ребенок способен отрицательно реагировать на пищу, поданную кусочками, или на комочки в блюде.
Постоянная тошнота провоцирует отказ от питания, а значит, потерю в весе.
Плохое сглатывание слюны, которая регулирует кислотность в желудке, и повышенное внутрибрюшное давление вызывают желудочно-пищеводный рефлюкс, то есть забрасывание содержимого желудка в пищевод. Это чревато такими последствиями, как воспаление стенки пищевода, респираторные инфекции.
Малоподвижный образ жизни, неврологические расстройства, неправильное питание провоцируют возникновение запоров у детей с синдромом Ретта. Они носят тяжелый характер, поскольку способны вызывать закупорку кишечника и сильные боли.
Повышенное слюнотечение, тошнота, рефлюкс снижают потребление ребенком пищи и даже развивают на нее негативную реакцию. В результате этого ребенок теряет в весе. Этот процесс стоит строго контролировать, поскольку он чреват истощением.
Другое тяжелое расстройство связано с работой дыхательной системы, развивающееся вплоть до приступов апноэ. Это явление настолько часто среди детей с синдромом, что нередко стает причиной их гибели.
Важными патогномоничными признаками синдрома считаются проявления аутизма. Именно из-за них заболевание изначально считали одной из форм этого расстройства, а в настоящее время относят к болезням аутистического спектра.
Аутистические признаки проявляются в отстранении от окружающего мира, в том числе и от родственников. Ребенок замыкается в себе, может не откликаться, когда его зовут. Предпочитает одиночество. Дети боятся чужих людей и непривычных ситуаций.
Лицо такого ребенка становится похожим на каменное. Взгляд блуждающий или устремлен в одну точку. Поведение часто непредсказуемо: случаются приступы неутомимого смеха или плача. Склонны к самоповреждениям: царапают кожу, кусают пальцы, вырывают волосы.
НЕТИПИЧНАЯ КАРТИНА
Наряду с типичной формой заболевания, описанной выше, встречаются и атипичные формы. Они имеют свои особенности, от которых зависит тяжесть заболевания.
- Zapella – форма синдрома с неярко выраженными признаками. Речь частично сохранена, умеренно выражен сколиоз, умственная отсталость средней степени тяжести. Физически развиваются нормально.
- Hanefeld – в клинической картине преобладает раннее развитие судорожных приступов. Часто они случаются даже до появления умственной деградации.
- Rolando – на первый план выходят признаки задержки психомоторного развития. Ребенок теряет возможность передвигаться, нарастает стереотипия движений, его беспокоят дыхательные нарушения.
Синдром Ретта – сложное генетическое заболевание. Прежде всего, его сопровождает полная умственная деградация и психоневрологические нарушения, влекущие за собой многочисленные патологии других систем организма.
К сожалению, в мире еще не существует способа кардинального искоренения болезни, хотя ученые ведут постоянные разработки в этом направлении.
Лечение синдрома сводится к трем основным направлениям. Медикаментозная терапия назначается для купирования судорожных припадков и стимуляции работы головного мозга.
Диетотерапия включает в себя контроль массы тела, употребление в пищу высококалорийных, витаминизированных продуктов.
Однако наибольшее внимание уделяется реабилитационным мероприятиям, направленным на укрепление опорно-двигательного аппарата и поддержание умственного, психомоторного развития.
Важно сохранить комплексный, всесторонний подход к проблеме. Такие дети нуждаются в постоянной поддержке со стороны взрослых и веры в них. Сотрудничество с ними, как с полноценной ячейкой общества, способствует их лучшей адаптации в социуме и более благоприятному развитию.
Синдром Ретта у детей – симптомы, лечение, прогноз
Синдром Ретта у детей – это редкое заболевание, поражающее почти исключительно девочек. Первые признаки выявляются в младенчестве, обычно в 4-5 месяцев жизни, и характеризуется умственной отсталостью и соматическими нарушениями. Патология встречается у одного ребенка на 10-15 тысяч новорожденных.

Причины
Синдром Ретта – что это такое? Ученые выяснили, что это генетическое заболевание. Причиной его появления является видоизменение особого гена МEСР2. Он контролирует синтез белков, которые необходимы для нормального построения нервной ткани и развития головного мозга.
Несколько фактов о синдроме Ретта у детей:
- мутация гена возникает во время внутриутробного развития, но первые признаки заболевания проявляются только через несколько месяцев после рождения;
- патология генетическая, но не наследственная – лишь у 1% наблюдается врожденный дефект гена по неизвестным причинам;
- некоторые женщины с мутантным геном нормально живут – их называют скрытыми носителями, поскольку болезнь находится в стадии компенсации.
Гипотеза о генетической этиологии подвергается критике, но на сегодняшний день это единственное научное объяснение причины возникновения синдрома Ретта.
Клинические признаки
Вначале у новорожденных симптомы могут совсем отсутствовать, иногда наблюдаются так называемые предвестники патологии. К ним относятся:
- бледность кожи;
- слабый тонус мышц;
- вялость конечностей;
- прохладная кожа;
- повышенная потливость.
Эти симптомы проявляются вследствие нарушения взаимосвязей между центральной нервной системой и вегетативными узлами и сплетениями, которые контролируют работу внутренних органов.
По мере роста ребенка органическое поражение головного мозга прогрессирует, появляются дополнительные признаки и неврологические симптомы:
- нарушение дыхания – оно частое и поверхностное, иногда может на несколько секунд останавливаться;
- ухудшение координации: однообразные движения, подергивания, ходьба с прямыми ногами;
- расстройства психики: гипервозбуждение, плаксивость, раздражительность, которые сменяются апатией и снижением настроения;
- нарушение роста: отставание в развитии, голова и конечности растут медленнее туловища;
- в старшем возрасте ребенок сильно отстает в развитии, наблюдаются серьезные сложности с обучением.
Иногда клиника сопровождается искривлением позвоночника, болями в спине, возникновением судорог. Для заболевания характерно постепенное начало и ухудшение состояния по мере прогрессирования системных расстройств.
Стадии заболевания
На основании клинической картины выделяют несколько стадий заболевания; они отражают прогрессирование патологии, а также имеют прогностическое значение в отношении возможной продолжительности жизни пациента. Если болезнь стремительно развивается, сопровождается выраженной неврологической симптоматикой, свидетельствующей об органическом поражение мозга, повышается риск внезапной смерти.
Различают четыре стадии заболевания:
- I – выявляется в раннем возрасте и характеризуется снижением тонуса мышц, нарушением роста костей и отсутствием других неврологических симптомов;
- II – к перечисленным признакам присоединяется нарушение походки, подергивание конечностей, увеличение размеров туловища;
- III – утрата некоторых привычных движений, затруднения речи, появление замкнутости;
- IV – серьезные речевые нарушения, появление тяжелых неврологических симптомов, сильная деформация позвоночника, укорочение конечностей.
Если у ребенка наблюдаются признаки четвертой стадии, прогноз будет неблагоприятным. Продолжительность жизни зависит от компенсации нарушений со стороны внутренних органов, которые поражаются при искривлении позвоночника и деформации грудной клетки.

Диагностика
Грамотно проведенное обследование – одно из условий эффективности лечения. Диагноз ставится на основании следующих критериев:
- осмотр – доктор проверяет тонус мускулатуры, рефлексы, проводит антропометрические измерения. Если наблюдается несоответствие размеров тела в раннем возрасте, диагноз синдрома Ретта ставится под вопросом;
- электроэнцефалография – отражает активность мозга. При выявлении серьезных нарушений уже выставляется диагноз без вопроса;
- УЗИ – необходимо для определения осложнений со стороны внутренних органов. Если ребенку больше 5 лет, можно провести МРТ для установления степени поражения головного мозга.
Лечение
Синдром Ретта является неизлечимой болезнью, вся терапия направлена на поддержку организма и компенсацию нарушений. Для этого применяются медикаменты, проводится коррекция образа жизни.
Лечение патологии включает в себя:
- прием ноотропных средств на основе пирацетама и пиритинола – для улучшения мозговой активности;
- назначение снотворных (барбитураты, мелатонин) – для снижения возбудимости и полноценного отдыха;
- применение препаратов против конвульсий на основе карбамазепина – при выраженных судорогах;
- препараты с ламотриджином – снижают выделение медиаторов в синаптическую щель, что отражается на частоте эпиприпадков;
- лечебную гимнастику – для сохранения мелкой моторики;
- работу с логопедом – с целью устранения речевой дисфункции;
- консультации психолога – для социальной адаптации.
Прогноз
Ответ на вопрос, сколько живут пациенты с синдромом Ретта, дать достаточно сложно. Если ребенок находится на поддерживающей терапии и соблюдает все рекомендации врача, прогноз благоприятный – при таких условиях можно прожить 40-50 лет. Очень важно принимать медикаменты, развивать умственные навыки и следовать всем предписаниям врача. Но у пациенток всегда есть риск остановки сердца.
Заключение
Синдром Ретта – неизлечимое генетическое заболевание, поражающее почти исключительно девочек. Патология характеризуется постепенным поражением головного мозга, развитием умственной отсталости с последующим нарушением роста костей и работы внутренних органов. Лечение направлено на компенсацию утраченных функций и замедление прогрессирования болезни.
Видео


* Представленная информация не может быть использована для самостоятельной постановки диагноза, определения лечения и не заменяет обращение к врачу!
Дети учат взрослых трем вещам: быть счастливым без причины, всегда быть чем-то занятым и уметь требовать изо всех сил то, чего хочется

