
Синдром Веста: малыш в беде
Синдром Веста — младенческая эпилепсия, описанная впервые еще в 19 веке. До открытия влияния адренокортикотропного гормона (АКТГ) на течение этого заболевание оно считалось неизлечимым.
История
В 1841 году английский педиатр Ульям Джеймс Вест (1793–1848) написал письмо главному редактору британского журнала The Lancet, где оно и было опубликовано. Письмо было озаглавлено: «О специфической форме младенческих пароксизмов». Непонятной болезнью с четырехмесячного возраста страдал сын Уильяма.
В письме доктор Вест описывал происходившие с сыном приступы как «наклоны». Ребенок наклонял голову до колен, а затем его тело полностью расслаблялось. Приступ мог длиться до 2–3 минут и включать до 20 «наклонов», интервалы между ними длились всего несколько секунд. Такие приступы доктор наблюдал у мальчика до 3 раз в день. В своем письме педиатр обращался к коллегам за помощью. В момент написания письма мальчику было около года, и он уже не мог приобретать новые навыки и не знал, как двигать конечностями, никогда не плакал и не смеялся, выглядел безучастным, не мог поддерживать тело в вертикальном положении и самостоятельно удерживать голову. К 11 месяцам приступы у мальчика стали напоминать генерализованные тонические.
В течение следующих 100 лет эпилептологи описывали схожие с описаниями Веста синдромы у детей, и к середине прошлого века в мировой литературе накопилось около 70 подобных случаев. В начале 60 х неврологи впервые описали ЭЭГ-паттерн у детей с пароксизмами: гипсаритмию, то есть беспорядочные высокоамплитудные несинхронные спайки и медленноволновую активность. В 1964 году впервые появился термин «синдром Веста».
Итак, что это за болезнь? Синдром Веста (СВ) — это эпилептическая энцефалопатия у детей, проявляющаяся триадой:
- Инфантильные спазмы (ИС). Это короткие сильные сокращения мышц, соединяющих голову с позвоночником, и мышц, расположенных вдоль позвоночника (сгибательные, разгибательные или смешанные).
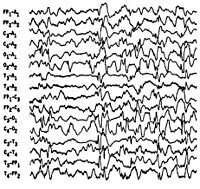
- Гипсаритмия — межприступные изменения на ЭЭГ.
- Прогрессирующее нарушение когнитивных, поведенческих и неврологических функций.
СВ встречается в 2–6 случаях на 10 000 новорожденных и составляет до 9 % эпилептических синдромов раннего детского возраста. От синдрома Веста чаще страдают мальчики — до 60 % от общего числа больных.
Формы
Официально СВ разделяют на симптоматическую (до 85 %), а также криптогенную и идиопатическую формы (вместе до 20 %). Но с клинической точки зрения у заболевания только 2 формы, так как различия между криптогенной и идиопатической формами практически отсутствуют. К симптоматической форме синдрома Веста относят случаи заболевания на фоне уже имеющейся патологии головного мозга или нарушений развития. У половины детей с симптоматической формой в анамнезе было осложненное течение внутриутробного периода: инфекции, метаболические расстройства, генетические и хромосомные дефекты (синдром Дауна и др.), а также нарушение внутриматочного кровообращения у матери. Реже наблюдается патология родового периода. Это гипоксически-ишемическое поражение мозга, травмы и другие осложнения в родах. К постнатальным причинам СВ относятся инфекции, травмы, гипоксически-ишемические инсульты и опухоли.
Криптогенную, или идиопатическую, форму заболевания диагностируют у детей с эпилепсией синдрома Веста без видимых причин, с нормальным психомоторным развитием и без повреждения головного мозга до возникновения заболевания. Это более благоприятная форма СВ.
Патогенез синдрома Веста в настоящее время неизвестен. У пациентов укорочена фаза REM-сна (фаза быстрого движения глаз), во время которой происходит нормализация ЭЭГ и снижение частоты спазмов. В связи с этим есть версия, что при СВ в стволе головного мозга имеет место дисфункция серотонинергических нейронов, участвующих в формировании циклов сна. Существуют и другие гипотезы, подразумевающие генетические и иммунные нарушения у маленьких пациентов.
Клиническая картина
Чаще всего синдром дебютирует у детей в возрасте 4–6 месяцев, причем более ранние симптомы— неблагоприятный прогностический фактор. Инфантильные спазмы синдрома Веста могут проявляться с высокой частотой и быть крайне разнообразными — сгибания туловища, вертикальные движения глазных яблок, или движения глаз, похожие на нистагм, а также «вскидывание» ручек по типу восточного приветствия и др. Один спазм длится доли секунды, спазмы группируются в серии — до 50 приступов в серии, количество серий в сутках — от одной до нескольких десятков. Часто приступы развиваются при пробуждении и засыпании, могут сопровождаться отведением головы или глаз в сторону. В спазм может быть вовлечена только половина тела. Появление приступов эпилепсии означает остановку психомоторного развития малыша, и часто — регресс приобретенных навыков. В 1–2 % случаев возможно спонтанное самоизлечение.
Лечение
Важнейшая задача терапии — полное прекращение или снижение частоты приступов и подавление гипсаритмии, которая делает невозможным нормальное развитие ребенка. Противоэпилептические средства в этом случае малоэффективны. Так возможно ли излечение от синдрома Веста?
В 1958 году в Европейском журнале о неврологии (European Journal of Neurology) была опубликована важнейшая работа по эпилепсии и эффективности введения кортикотропина при инфантильных спазмах (авторы Л. Сорель и A. A. Дюшан-Бойоль). АКТГ помогал в 50–90 % случаев, причем лечению лучше поддавалась криптогенная форма, чем симптоматическая. В большом финском исследовании 1980 года летальные осложнения при терапии кортикотропином достигали 5 %, а частота серьезных побочных эффектов составила 37 %. Высокий риск осложнений и низкая эффективность кортикотропина при симптоматической форме СВ привели к необходимости дальнейшего поиска препаратов для купирования спазмов.
Сейчас используются и другие гормональные средства — преднизолон, дексаметазон и тетракозактид. Последний препарат — это синтетический полипептид, обладающий свойствами эндогенного кортикотропина и дающий меньше осложнений, чем сам кортикотропин. В течение последних 20 лет зарекомендовал себя противоэпилептический препарат вигабатрин. Восприимчивость к терапии составляет 23–68 %. До сих пор не определены оптимальные дозы и продолжительность лечения ни для вигабатрина, ни для кортикотропина и тетракозактида.
Кроме того, при лечении синдрома Веста назначают вальпроаты и бензодиазепины. Однако полное исчезновение инфантильных спазмов на фоне приема этих препаратов наступает позже, чем при лечении стероидами и вигабатрином. При локализованном очаге эпилептоидной активности возможно хирургическое лечение, однако эффективно оно далеко не во всех случаях.
Динамика обязательно оценивается ЭЭГ-мониторингом, поскольку на фоне терапии спазмы могут перейти в субклинические, которые трудно распознать без ЭЭГ. В ремиссии (месяц без приступов) гипсаритмия может полностью исчезнуть, сменяясь нормальным вариантом ЭЭГ. Но в 23–50 % случаев синдрома Веста прогноз не очень хороший — заболевание трансформируется в другие формы эпилепсии, которые иногда могут проявить себя только в пубертатном периоде.
Прогноз
Со времени письма Уильяма Веста в The Lancet прогноз для «вестиков», несомненно, улучшился, но всё еще остается крайне серьезным. К сожалению, летальность от самого заболевания или осложнений его лечения в течение первых 3 лет жизни доходит до 11 % и за последние 40 лет она остается неизменной. Нормальное интеллектуальное развитие сохраняется у 9–28 % детей. Нормальный или близкий к нормальному интеллект при криптогенной и идиопатической формах сохраняется чаще — в 38–78 % случаях, тогда как при симптоматической форме — только у 2–18 % детей. Прогноз каждого ребенка с синдромом Веста крайне индивидуален — качество и продолжительность жизни зависит как от формы самого заболевания, так и от своевременности и эффективности лечения.
Синдром Веста : причины, симптомы, диагностика, лечение
В таблице ниже перечислены основные причины синдрома Веста. Две основные группы — симптоматические и криптогенные спазмы. Криптогенные случаи определяются по-разному: как те, при которых невозможно выявить вызывающую их причину или при которых ребенок развивается нормально до начала приступов (Lombroso, 1983а).
Определение криптогенный не исключает наличия причины, таким образом, различие между этими двумя группами размываются. В соответствии с последним приведенным определением криптогенные спазмы составляют 10-15% случаев, но их течение, вероятно, будет более благоприятным, чем симптоматических. Некоторые исследователи (Vigevano et al., 1993; Dulac и Tuxhorn, 2005) описывают идиопатическую группу инфантильных спазмов с хорошим прогнозом и характерными клиническими и ЭЭГ признаками.
Однако Haga et al. (1995) и Watanabe et al. (2001) не удалось дифференцировать этиологическую группу только на основании клинических и ЭЭГ (иктальных и интериктальных) признаков. Симптоматические спазмы чаще всего развиваются при дизгенезе головного мозга и нейрокутанных синдромах (чаще всего туберозном склерозе) (Vigevano et al., 1994а; Arzimanoglou et al., 2004; Dulac и Tuxhorn, 2005). Выраженные мальформации включают в себя лиссэнцефалию, пахигирию, диффузные субкортикальные гетеротопии и агенезию мозолистого тела, особенно синдром Айкарди.
Туберозный склероз вызывает 10-20% случаев. Фокальные кортикальные дисплазии (Chugani et al., 1990, 1996; Robain и Vinters, 1994; Chugani и Conti, 1996), вероятно, являются отдельной наиболее важной причиной инфантильных спазмов. Хронические врожденные и приобретенные поражения являются причиной в большинстве остальных симптоматических случаев (Cowan и Hudson, 1991). Инфантильные спазмы также наблюдаются при связанных с эпилепсией хромосомных нарушениях (Battaglia и Guerrini, 2005).
Синдром Веста обычно возникает спорадически, за исключением тех случаев, когда он связан с метаболическими расстройствами. Генетически детерминированные формы редки. Случаи, связанные с аномалиями Х-хромосомы, в основном вызваны мутациями гена ARX (Suri, 2005) и могут сочетаться с другими, в том числе врожденными, аномалиями.
Также сообщалось о случаях, вызванных мутациями хромосомы Xp11-Xpter (Claes et al., 1997) и гена CDLK5 (Weaving et al., 2004). Также спазмы, вместе с периферическими отеками, гипсаритмией и атрофией зрительного нерва, являются частью синдрома РЕНО. С патологоанатомической точки зрения, большинство причин инфантильных спазмов, выявленных на вскрытии, были связаны с пренатальными факторами. Meencke и Gerhard (1985) не выявили патологических изменений только в 11% из 107 случаев.
Большинство случаев вызывается диффузными поражениями, но и односторонние нарушения, особенно порэнцефалическая киста в бассейне средней мозговой артерии (Palm et al., 1988), могут вызывать инфантильные спазмы, прогноз которых может быть и благоприятным (Alvarez et al., 1987; Cusmai et al., 1988).
He существует общепринятого подхода к лечению синдрома Веста. Большинство современных антиконвульсантов неэффективны. До настоящего времени в последних обзорах, таких как обзор Cochrane (Hancock et al., 2003) и рекомендациях Американской академии практической неврологии (Mackay et al., 2004) признаются эффективными только кортикостероиды или АКТГ и вигабатрин. Некоторые авторы признают эффективность других препаратов, например, топирамата, ламотриджина, вальпроата, пиридоксина и зонисамида, и нефармакологических методов (подробнее см. Arzimanoglou et al., 2004; Dulac и Tuxhorn, 2005).
Во всех работах подчеркивается необходимость проведения хорошо организованных проспективных исследований с большим числом участников, длительным периодом наблюдения, проводимых с учетом этиологии.

а) Гормональная терапия. Применяются как АКТГ, так и кортикостероиды. Схемы применения АКТГ крайне вариабельны. Дозы АКТГ варьируют от 10 до 40 единиц природного гормона или от 0,1 до 0,5 мг/кг тетракосактида. Кортикостероиды, включая преднизолон 2-10 мг/кг, гидрокортизон 5-20 мг/кг/день, и дексаметазон 0,3-0,5 мг/кг/день применяются в виде монотерапии или в различных комбинациях с АКТГ. Проведено всего несколько контролируемых исследований, и еще меньше сравнительных исследований (Glaze et al., 1988; Mackay et al., 2004).
По данным одного контролируемого сравнительного исследования тетракосактида в сравнении с предни-золоном, проведенного на небольшой группе пациентов, АКТГ оказался более эффективным, вызвав ремиссию у 13 из 15 младенцев, по сравнению с 4 из 14, получавших преднизолон (Baram et al., 1996). Наоборот, в недавнем большом контролируемом исследовании инфантильных спазмов в Соединенном Королевстве (United Kingdom Infantile Spasms Study — UKISS, Lux et al., 2005) не было выявлено преимущества одного препарата перед другим.
В другом исследовании применение АКТГ в высоких дозах (150 МЕ/кг/день) оказалось не более эффективным, чем в низких (20 МЕ/кг/день) (Hrachovy et al., 1994), и результаты применения тетракосактида в низких дозах по двум разным схемам не отличались в группах, получавших препарат в более низкой и более высокой дозировке (Kondo et al., 2005). В настоящее время опубликованы и другие отчеты (обзор см. Arzimanoglou et al., 2004). Heiskala et al. (1996) предложили индивидуализировать прогрессивную терапию АКТГ, начинать лечение с дозы 3 МЕ/кг/день и при отсутствии эффекта прогрессивно повышать ее до максимальной дозы 12 МЕ/кг/день. На основании клинического опыта складывается впечатление, что отдельные случаи могут оказаться чувствительны к применению какого-либо из названных препаратов.
Применение кортикостероидов не требует выполнения инъекций и, таким образом, укорачивает срок госпитализации, многие клиницисты по-прежнему используют их именно по этой причине. Однако АКТГ может выйти на первое место, если более убедительные крупные исследования подтвердят его преимущества. Механизм действия обоих препаратов остается неизвестным. Длительность курса терапии не менее вариабельна, от 3-4 недель до нескольких месяцев. Мы в своей практике проводим гормональную терапию короткими курсами в пределах 4-6 недель.
Терапия АКТГ и стероидами позволяет контролировать спазмы в 50-70% случаев и нормализует или улучшает ЭЭГ у несколько меньшей части больных. Рецидив возникает у 25-30% пациентов. Долгосрочные эффекты не столь однозначны (Riikonen 1996; Gaily et al., 1999; Goh et al., 2005); нормальное развитие когнитивных функций наблюдается только у 25-30% пациентов, еще у 10-15% имеются легкие нарушения; при криптогенных случаях вероятность благоприятного исхода больше, она составляет 50% и 25% соответственно. Припадки наблюдаются примерно у половины пациентов. Обычно они соответствуют картине синдрома Леннокса-Гасто, но также наблюдаются и парциальные припадки.
Побочные эффекты применения АКТГ или кортикостероидов развиваются часто и могут протекать тяжело, они включают гипертензию, инфекции и метаболические расстройства. Помимо классических побочных эффектов терапии кортикостероидами, часто наблюдается атрофия головного мозга (Konishi et al., 1992). Следовательно, перед началом терапии необходима нейровизуализация головного мозга.
б) Антиэпилептические препараты. В нескольких исследованиях была выявлена эффективность вигабатрина (подробнее см. Arzimanoglou et al., 2004; Dulac и Tuxhorn, 2005). В дозировке 100-150 мг/день он начинает контролировать спазмы приблизительно в 50-60% случаев обычно менее чем за неделю (Aicardi et al., 1996). Его воздействие на ЭЭГ-картину развивается медленнее, чем у кортикостероидов. В сравнительном исследовании при применении гормонального лечения как терапии первой линии (Vigevano и Cilio, 1997) вигабатрин оказался более эффективным при лечении инфантильных спазмов, вызванных церебральными мальформациями или туберозным склерозом, тогда как АКТГ показал большую эффективность в случаях, причиной которых были гипоксически-ишемические поражения. При криптогенных случаях эффективность обоих препаратов оказалось одинаковой.
Исчезновение нарушений на ЭЭГ у пациентов, случайным образом получавших АКТГ, наблюдалось раньше, чем у тех, кто получал первичную терапию вигабатрином.
В мультицентровом рандомизированном контролируемом исследовании UKISS (O’Callaghan et al., 2004; Lux et al., 2005; Riikonen, 2005) на 13 и 14 день лечения спазмы прекратились у 40 пациентов (73%) из 55, получавших гормональную терапию (преднизолон 21/30, тетракосактид 19/25), и 28 (54%) из 52 детей, получавших вигабатрин (разница 19%, 95% доверительный интервал (ДИ) 1-36%, р=0,043). Два младенца, которым был назначен тетракосактид и один, которому был назначен вигабатрин, получали преднизолон. Побочные эффекты зафиксированы у 30 (55%) из 55 детей, получавших гормональную терапию, и у 28 (54%) из 52 младенцев, получавших вигабатрин.
При обследовании тех же пациентов в возрасте 12-14 месяцев (Lux et al., 2005) отсутствие спазмов наблюдалось у одинакового числа пациентов в обеих группах: гормоны 41/55 (75%), вигабатрин 39/51 (76%). В этом исследовании также проводилась оценка нервного развития, для этих целей применялась Шкала адаптивного поведения Вайнленд (Vineland Adaptive Behavior Scales — VABS). Средний уровень развития различался незначительно: гормоны 78,6 (С016,8) в сравнении с вигабатрином 77,5 (СО12,7), за исключением группы младенцев, у которых этиология заболевания не была установлена: в этой группе средний уровень развития по шкале VABS у получавших гормональную терапию был выше, чем у лечившихся вигабатрином.
Вигабатрин показывает высокую эффективность при спазмах, вызванных туберозным склерозом (Chiron et al., 1990, 1997) и в настоящее время является препаратом выбора в этих случаях (Aicardi et al., 1996), тогда как гормональная терапия рекомендована некоторыми авторами (Riikonen, 2005) в криптогенных случаях. Однако трактовка выявленных различий вызывает трудности, и названные авторы рекомендуют вигабатрин для первичного применения из-за его быстрого эффекта (часто его воздействие на припадки проявляется менее чем за неделю) и, в целом, хорошую переносимость препарата. Рекомендуемая доза в 100-150 мг/кг/день. В случаях неэффективности вигабатрина в течение нескольких дней следующим шагом является пробное назначение стероидов (Arzimanoglou et al., 2004).
Самый серьезный побочный эффект вигабатрина — это сужение полей зрения, которое наблюдается у 30-50% взрослых пациентов и может не регрессировать. Его частота значительно варьирует в различных исследованиях. Vanhatalo et al. (2002) обнаружил, что у 17 (18,5%) из 91 ребенка, получавшего препарат, развивалось сужение поля зрения с височной стороны. Присутствует положительная корреляция между степенью сужения поля зрения и длительностью лечения и дозой препарата. Самый короткий курс вигабатрина, вызывавший сужение полей зрения, составил 15 месяцев, наименьшая общая доза — 914 г.
Hammoundi et al. (2005) при наблюдении группы из 28 детей выявил снижение контрастной чувствительности и остроты зрения. Обычно это нарушения легкой степени, и маловероятно, что лечение короткими курсами (менее шести месяцев) вызовет тяжелые нарушения; поэтому мы считаем, что соотношение рисков при таком тяжелом состоянии, как инфантильные спазмы, свидетельствует в пользу применения этого препарата.
Рецидивы как при гормональной терапии, так и при лечении вигабатрином, случаются нередко, и еще предстоит установить оптимальную продолжительность лечения.
Другие методы лечения инфантильных спазмов включают применение бензодиазепинов, особенно нитразепама. Они иногда бывают эффективными в отношении клинических спазмов (Chamberlain, 1996), но оказывают ограниченное воздействие на изменения ЭЭГ. В одном сравнительном исследовании не было выявлено различий между применением нитразепама и гормональной терапией (Dreifuss et al., 1986), но большинство авторов считает бензодиазепины менее эффективными, чем стероиды.
Вальпроат натрия в высоких дозах (100-200 мг/кг/день), по данным некоторых исследователей (Siemes et al., 1988; Prats et al., 1991; Ohtsuka et al., 1994), может давать хорошие результаты, позволяя контролировать спазмы в 40-65% случаев.
Прогноз инфантильных спазмов при лечении АКТГ/стероидами или вигабатрином зависит от этиологии заболевания; наилучшие результаты получены при криптогенных (или идиопатических) случаях с совершенно нормальным развитием до начала спазмов. Наличие в анамнезе припадков, неврологической симптоматики и значительных отклонений на МРТ являются неблагоприятными прогностическими факторами. В исследовании Коо et al. (1993) средний коэффициент развития у 17 больных с криптогенными спазмами составил 71, у 40 пациентов с симптоматическими спазмами — 48. У 51% пациентов были припадки других типов, это свидетельствовало о симптоматической природе заболевания и являлось плохим прогностическим признаком для когнитивного развития.
Качественная оценка влияния выбранной терапии на общее развитие ребенка не производилась. Одна из основных трудностей проведения подобных исследований заключается в том, что причиной синдрома Веста могут являться различные факторы.
г) Хирургическая резекция очагов дисплазии головного мозга, считающихся ответственными за развитие спазмов (Chugani et al, 1990; Shields et al., 1992; Chugani и Pinard, 1994; Jonas et al., 2005; Kang et al., 2006) может быть показана в отдельных случаях в начале лечения, особенно когда на МРТ четко выявлено диспластическое поражение или в случаях гемимегэнцефалии.
Границы поражения коры обычно определяются при МРТ, но в некоторых случаях их можно выявить только при позитронной эмиссионной томографии (ПЭТ — Chugani et al., 1996) или фотонной эмиссионной компьютерной томографии (Miyazaki et al., 1994). Однако были обнаружены транзиторные аномалии при ПЭТ, особенно в период активной гипсаритмии (Watanabe et al., 1994); взаимоотношения между аномалиями, выявленными при ПЭТ, причинами и исходом заболевания пока неясны (Watanabe, 1996). Предпринимались попытки выделения группы младенцев с проблемами в неонатальном периоде и повышенным риском развития инфантильных спазмов и проведения превентивного лечения (Okumura и Watanabe, 2001).
Синдром Веста
Синдром Веста — серийные спастические сокращения в отдельных мышечных группах или генерализованного характера, протекающие на фоне задержки нейропсихического развития и сопровождающиеся гипсаритмическим ЭЭГ-паттерном. Манифестирует в возрасте до 4-х лет, преимущественно на 1-ом году жизни. В большинстве случаев имеет симптоматический характер. Диагностика синдрома основана на клинических данных и результатах ЭЭГ. Для выявления основной патологии необходимы КТ или МРТ, ПЭТ головного мозга, консультация генетика, нейрохирурга. Лечение возможно противоэпилептическими препаратами, стероидами (АКТГ, преднизолон), вигабатрином. По показаниям решается вопрос о хирургическом лечении (каллозотомия, удаление патологического очага).
Общие сведения
Синдром Веста носит название по имени врача, наблюдавшего его проявления у своего ребенка и впервые описавшего его в 1841 г. В связи с манифестацией синдрома в раннем возрасте и протеканием судорог по типу серии отдельных спазмов, пароксизмы, характеризующие синдром Веста, получили название инфантильные спазмы. Первоначально заболевание относили к генерализованной эпилепсии. В 1952 г. был изучен специфический гипсаритмический ЭЭГ-паттерн, патогномоничный для этой формы эпилепсии и характеризующийся медленноволновой асинхронной активностью с беспорядочными спайками высокой амплитуды. В 1964 г. специалистами в области неврологии синдром Веста был выделен в качестве отдельной нозологии.
Внедрение в неврологическую практику нейровизуализации позволило определить наличие у пациентов очаговых поражений вещества мозга. Это заставило неврологов пересмотреть свои взгляды на синдром Веста как на генерализованную эпилепсию и отнести его в ряд эпилептических энцефалопатий. В 1984 г. был выявлена эволюция эпилептической формы энцефалопатии от ее раннего варианта в синдром Веста, а с течением времени в синдром Леннокса-Гасто.
В настоящее время синдром Веста занимает около 2% от всех случаев эпилепсии у детей и примерно четверть младенческой эпилепсии. Распространенность составляет, по различным источникам, от 2 до 4,5 случаев на 10 тыс. новорожденных. Несколько чаще заболевают мальчики (60%). 90% случаев манифестации синдрома приходится на 1-й год жизни, с пиком в возрасте от 4 до 6 мес. Как правило, к возрасту 3-х лет мышечные спазмы проходят или трансформируются в иные формы эпилепсии.
Причины синдрома Веста
В подавляющем большинстве случаев синдром Веста носит симптоматический характер. Он может возникать вследствие перенесенных внутриутробных инфекций (цитомегалии, герпетической инфекции), постнатального энцефалита, гипоксии плода, преждевременных родов, внутричерепной родовой травмы, асфиксии новорожденного, постнатальной ишемии вследствие позднего пережатия пуповины. Синдром Веста может являться следствием аномалий строения головного мозга: септальной дисплазии, гемимегалоэнцефалии, агенезии мозолистого тела и пр. В ряде случаев инфантильные спазмы выступают симптомом факоматозов (синдрома недержания пигмента, туберозного склероза, нейрофиброматоза), точечных генных мутаций или хромосомных аберраций (в т. ч. синдрома Дауна). В литературе упоминаются случаи фенилкетонурии с инфантильными спазмами.
В 9-15% синдром Веста является идиопатическим или криптогенным, т. е. его первопричина не установлена или не очевидна. Зачастую при этом прослеживается наличие случаев фибрильных судорог или эпиприступов в семейном анамнезе больного ребенка, т. е. имеет место наследственная предрасположенность. Ряд исследователей указывают, что фактором, провоцирующим синдром Веста, может выступать вакцинация, в частности введение АКДС. Это может быть связано с совпадением сроков вакцинации и возраста типичного дебюта синдрома. Однако достоверные данные, подтверждающие провоцирующую роль вакцин, до сих пор получены.
Патогенетические механизмы возникновения инфантильных спазмов являются предметом изучения. Существует несколько гипотез. Одна из них связывает синдром Веста с расстройством функционирования серотонинергических нейронов. Действительно, у пациентов наблюдается понижение уровня серотонина и его метаболитов. Но пока неизвестно, является оно первичным или вторичным. Обсуждалась также иммунологическая теория, связывающая синдром Веста с увеличением количества активированных В-клеток. Положительный лечебный эффект АКТГ лег в основу гипотезы о сбоях в системе «мозг-надпочечники». Отдельные исследователи предполагают, что в основе синдрома лежит избыточное количество (гиперэкспрессия) возбуждающих синапсов и проводящих коллатералей, формирующих повышенную возбудимость коры. Асинхронность ЭЭГ-паттерна они связывают с физиологичным для этого возрастного периода недостатком миелина. По мере созревания мозга происходит уменьшение его возбудимости и нарастание миелинизации, что объясняет дальнейшее исчезновение пароксизмов или их трансформацию в синдром Леннокса-Гасто.
Симптомы синдрома Веста
Как правило, симптом Веста дебютирует на первом году жизни. В отдельных случаях его манифестация происходит в более старшем возрасте, однако не позже 4-х лет. Основу клиники составляют серийные мышечные спазмы и нарушение психомоторного развития. Первые пароксизмы зачастую появляются на фоне уже существующей задержки психомоторного развития (ЗПР), но в 1/3 случаев возникают у первично здоровых детей. Отклонения в нейропсихологическом развитии наиболее часто проявляются снижением и выпадением хватательного рефлекса, аксональной гипотонией. Возможно отсутствие слежения глазами за предметами и расстройство фиксации взора, что является прогностически неблагоприятным критерием.
Мышечные спазмы носят внезапный симметричный и кратковременный характер. Типична их серийность, при этом интервал между следующими друг за другом спазмами длится не менее 1 минуты. Обычно наблюдается возрастание интенсивности спазмов в начале пароксизма и ее спад в конце. Число спазмов, происходящих за сутки, варьирует от единиц до сотен. Наиболее часто возникновение инфантильных спазмов происходит в период засыпания или сразу после сна. Провоцировать пароксизм способны резкие громкие звуки и тактильная стимуляция.
Семиотика пароксизмов, которыми сопровождается синдром Веста, зависит от того, какая мышечная группа сокращается — экстензорная (разгибательная) или флексорная (сгибательная). По этому признаку спазмы классифицируют на экстензорные, флексорные и смешанные. Чаще всего наблюдаются смешанные спазмы, затем сгибательные, наиболее редко — разгибательные. В большинстве случаев у одного ребенка наблюдаются спазмы нескольких видов и то, какой именно спазм будет преобладать, зависит от положения тела в момент начала пароксизма.
Может иметь место генерализованное сокращение всех мышечных групп. Но более часто наблюдаются локальные спазмы. Так, судороги в сгибателях шеи сопровождаются кивками головой, спазмы в мускулатуре плечевого пояса напоминают пожимание плечами. Типичным является пароксизм по типу «складного ножа», обусловленный сокращением мышц сгибателей живота. При этом тело как бы складывается пополам. Инфантильные спазмы верхних конечностей проявляются отведением и приведением рук к туловищу; со стороны кажется, что ребенок сам себя обнимает. Сочетание подобных спазмов с пароксизмом по типу «складного ножа» ассоциируется с принятым на Востоке приветствием «салаам», поэтому было названо «салаамовой атакой». У детей, которые умеют ходить, спазмы могут протекать по типу дроп-атак — неожиданных падений с сохранением сознания.
Наряду с серийными спазмами, синдром Веста может сопровождаться бессудорожными приступами, проявляющимися внезапной остановкой двигательной активности. Иногда отмечаются пароксизмы, ограниченные подергиванием глазных яблок. Возможно нарушение дыхания вследствие спазма дыхательной мускулатуры. В некоторых случаях имеют место асимметричные спазмы, проявляющиеся отведением головы и глаз в сторону. Могут встречаться и другие виды эпиприступов: фокальные и клонические. Они комбинируются со спазмами или имеют самостоятельный характер.
Диагностика синдрома Веста
Синдром Веста диагностируется по основной триаде признаков: приступы кластерных мышечных спазмов, задержка психомоторного развития и гипсаритмический ЭЭГ-паттерн. Имеют значение возраст манифестации спазмов и их связь со сном. Трудности диагностики возникают при позднем дебюте синдрома. В ходе диагностики ребенок консультируется педиатром, детским неврологом, эпилептологом, генетиком. Дифференцировать синдром Веста следует с доброкачественным младенческим миоклонусом, доброкачественной роландической эпилепсией, младенческой миоклонической эпилепсией, синдромом Сандифера (наклон головы по типу кривошеи, гастроэзофагальный рефлюкс, эпизоды опистотонуса, которые могут быть приняты за спазмы).
Интериктальная (межприступная) ЭЭГ характеризуется наличием дезорганизованной беспорядочной, динамично изменяющейся спайк-волновой активности, как в период бодрствования, так и во сне. Проведение полисомнографии позволяет выявить отсутствие спайк-активности в период глубоких стадий сна. Гипсаритмия регистрируется в 66% случаев, обычно на ранних стадиях. Позже наблюдается некоторая организация хаотичного ЭЭГ-паттерна, а в возрасте 2-4 лет его переход в комплексы «острая-медленная волна». Наиболее частый иктальный ЭЭГ-паттерн (т. е. ЭЭГ-ритм в период спазмов) — это генерализованные медленноволновые комплексы высокой амплитуды с последующим угнетением активности не менее 1 сек. При регистрации на ЭЭГ фокальных изменений следует думать об очаговом характере поражения головного мозга или наличии аномалий его строения.
КТ головного мозга у имеющих синдром Веста детей может выявлять диффузные либо очаговые изменения церебральных структур, но может быть в пределах нормы. В диагностике локальных поражений более чувствительным методом является МРТ головного мозга. Для выявления участков гипометаболизма мозговых тканей в некоторых случаях возможно проведение ПЭТ головного мозга.
Лечение синдрома Веста
Синдром Веста считался резистентным к проводимой терапии вплоть до открытия в 1958 г. влияния на приступы препаратов АКТГ. Терапия АКТГ и преднизолоном приводит к значительному улучшению или полному прекращению инфантильных спазмов, что сопровождается исчезновением гипсаритмического ЭЭГ-паттерна. До сих пор среди неврологов нет однозначных решений касательно доз и длительности стероидной терапии. Исследования показали, что в 90% случаев терапевтический успех достигался при применении больших дозировок АКТГ. Сроки терапии могут варьировать в пределах 2-6 недель.
Новый этап в лечении инфантильных спазмов начался в 1990-1992 гг. после обнаружения положительного терапевтического эффекта вигабатрина. Однако преимущество лечения вигабатрином пока доказано лишь для больных туберозным склерозом. В остальных случаях исследования показали большую эффективность стероидов. С другой стороны стероидная терапия имеет худшую, в сравнении с вигабатрином, переносимость и более высокий процент рецидивов.
Из антиконвульсантов эффективность показана лишь у нитразепама и вальпроевой кислоты. У отдельных пациентов описан лечебный эффект больших доз витамина В6, который отмечался в первые недели терапии. При инфантильных спазмах, резистентных к проводимой терапии, с подтвержденным на томографии наличием патологического очага показана консультация нейрохирурга для решения вопроса о резекции очага. Если подобная операция невозможна, то при наличии дроп-атак проводится тотальная каллозотомия (пересечение мозолистого тела).
Прогноз синдрома Веста
Обычно к 3-летнему возрасту наблюдается регресс и исчезновение инфантильных спазмов. Но примерно в 55-60% случаев они трансформируются в другую форму эпилепсии, чаще всего в синдром Леннокса-Гасто. Фармакорезистентность часто констатируется при инфантильных спазмах, сопровождающих синдром Дауна. Даже при успешном купировании пароксизмов синдром Веста имеет неудовлетворительный прогноз в плане психомоторного развития ребенка. Возможны когнитивные и поведенческие нарушения, ДЦП, аутизм, трудности в обучении. Остаточный психомоторный дефицит не наблюдается только в 5-12% случаев. ЗПР отмечается у 70-78% детей, двигательные расстройства — у 50%. Серьезный прогноз имеет синдром Веста, обусловленный аномалиями или дегенеративными изменениями головного мозга. При этом летальность может достигать 25%.
Более благоприятный прогноз имеют криптогенный и идиопатический синдром Веста при отсутствии ЗПР до появления спазмов. В этой группе больных остаточный интеллектуальный или неврологический дефицит отсутствует у 37-44% детей. Неблагоприятно отражается на прогнозе болезни откладывание начала лечения. Прогностическая оценка затрудняется тем, что отдаленные последствия также зависят от основной патологии, на фоне которой возникает симптоматический синдром Веста.
Синдром Веста. Как лечить? Ответ детского эпилептолога.
Синдром Веста – это возрастзависимый эпилептический синдром, относящийся к группе младенческих эпилептических энцефалопатий и характеризующийся особым видом приступов – инфантильными спазмами.
Синдром впервые был описан в 1841 году английским доктором Вестом, когда данное состояние наблюдалось у его ребёнка.
Есть несколько видов синдрома Веста: симптоматический, криптогенный и идиопатический. У каждого вида есть свои характерные ему особенности, течение заболевания и свой прогноз на будущее.
При данном виде заболевания очень важно провести раннюю диагностику, определить диагноз и начать соответствующее лечение, чтобы предотвратить тяжелые когнитивные нарушения. В большинстве случаев, заболевание проявляется у детей в возрасте нескольких месяцев.
Симптомы синдрома Веста
Как правило, синдром проявляется в первые месяцы жизни или в течение первого года жизни ребёнка (до года).
Основным симптомом заболевания являются приступы в виде спазмов. Во время приступа у ребёнка сокращаются различные группы мышц тела и конечностей. Приступы носят серийный характер и, в большинстве случаев, длятся до 15 минут.
Инфантильные спазмы всегда начинаются непредсказуемо и могут возникнуть в любое время суток, но обычно наблюдаются во время пробуждения или засыпания ребёнка. В некоторые случаях, могут наблюдаться генерализованные сокращения мышц, но наиболее частыми являются локальные спазмы.

Причины синдрома Веста
В большинстве случаев, синдром носит симптоматический характер и его причиной является поражение головного мозга по какой-либо причине. Самой распространённой причиной заболевания у детей является гипоксия при родах. Гипоксия – это кислородное голодание головного мозга, которое может возникнуть из-за патологических состояний, при неблагоприятном течении беременности или осложнениях при родах, например, обвитие пуповиной шеи плода.
Помимо этого, к заболеванию могут привести:
- инфекционные заболевания головного мозга в период после рождения;
- структурные патологии головного мозга;
- туберозный склероз;
- генетические и метаболические нарушения;
- структурные изменения в головном мозге;
- ряд других причин.
Внимание родителей! Если причина появления синдрома у вашего ребёнка не обнаружена, крайне важно провести генетические тесты перед планированием следующего ребёнка.
Диагностика
- ЭЭГ – данное исследование крайне важно во время диагностики синдрома Веста, так как на электроэнцефалограмме можно будет заметить определенную, специфическую для заболевания картину.
- МРТ – необходимо для определения наличия поражений головного мозга. В маленьком возрасте данное исследование проводится под наркозом.
- Генетические тесты и метаболические исследования – помогут дать больше информации о вашем случае и подтолкнуть к более эффективному протоколу лечения.
- Дополнительные проверки – они могут быть назначены при необходимости исключения наличия иных заболеваний (о них мы не будет говорить в данной статье, так как это достаточно объемная тема).
ВАЖНО! При первых появлениях настораживающих или подозрительных движений у младенца, следует как можно скорее проконсультироваться с педиатром и неврологом. Обратите внимание, если у малыша наблюдаются регулярные, серийные, цикличные движения. Желательно не ждать очереди даже пару недель!
Иногда родители обращаются за помощью не сразу, особенно если инфантильные спазмы не происходили сериями, что приводит к необративным последствиям.
Лечение синдрома Веста
В большинстве случаев, синдром Веста у детей требуется агрессивное медикаментозное лечение, чтобы предотвратить дальнейшие когнитивные нарушения у ребенка.
Наиболее эффективными противоэпилептическими препаратами при синдроме Веста считаются Сабрил и стероиды в больших дозировках.
Как показывает опыт наших иностранных пациентов, во многих странах мира, в том числе в России, Украине, Казахстане и других странах бывшего СССР, клинические рекомендации могут не включать достаточно высокие дозировки из-за тяжелых побочных эффектов, что, к сожалению, может не дать нужного терапевтического эффекта.
Прогноз
К нам часто поступает вопрос: можно ло ли вылечить синдром Веста и каков прогноз на будущее? Смотрите статистику и факты.
- Примерно у половины детей эпилептические приступы прекратятся.
- В плане моторики у большинства детей не будет проблемы с развитием.
- Симптоматический и криптогенный виды синдрома Веста часто сопровождаются определенной задержкой когнитивного развитии, причем во многих случаях на ранних стадиях тяжело определить тяжесть поражения или его наличие, что затрудняет прогноз.
- У каждого пятого ребёнка с синдромом Веста после наблюдается синдром Леннокса-Гасто.
- В большинстве случаев, дети с данным синдромом страдают от фармакорезистентной эпилепсии, устойчивой к медикаментозной терапии.
Хотите узнать больше о синдроме Веста и необходимом вашему ребёнку лечении?
Подробную информацию о синдроме Веста, диагностике и протоколах лечения можно найти в книге профессора Ури Крамера “Детская эпилепсия от А до Я”. Профессор даёт четкие рекомендации родителям на что нужно обратить внимание на том или ином этапе лечения, а также даёт прогноз развития заболевания в будущем.
Хотите знать, что действительно помогает при эпилепсии и как помочь ребёнку жить максимально полноценной жизнью, несмотря на заболевание?
Раз в неделю мы выпускаем видео или статью о лечении эпилепсии. Это БЕСПЛАТНАЯ и ЕДИНСТВЕННАЯ в своем роде электронная рассылка в мире и мы уверены, что в этих выпусках вы найдёте много полезных рекомендаций для себя и своего ребёнка.
Первый выпуск, который Вы получите – “9 Главных вопросов и ответов об эпилепсии”.

- Можно ли вылечить эпилепсию у детей?
- Переходит ли эпилепсия по наследству?
- Помогают ли альтернативные методы лечения контролировать приступы?
- Что нельзя делать при эпилепсии?
- Сколько должен спать ребёнок с эпилепсией?
- Чем опасны приступы эпилепсии во сне?
- Как помочь пациенту во время приступа?
- Можно ли заниматься спортом?
- Может ли эпилепсия привести к проблемам в учебе, задержке развития, проблем с памятью и поведением?
Введите адрес вашей электронной почты и проверьте почту через 5 минут
***Мы ценим вас и ваше доверие. Наша цель – предоставить вам достоверную информацию о лечении эпилепсии, а также постараться помочь вам или вашим детям жить с этим тяжелым заболеванием. Ни при каких обстоятельствах ваши данные не будут переданы и проданы третьим лицам. Как и вы, мы не любим получать бесполезную почту или рекламу, и постараемся оправдать ваше доверие.
Читайте книгу детского эпилептолога профессора Ури Крамера «Детская эпилепсия от А до Я»

Узнайте, как помочь ребёнку жить максимально возможной полноценной жизнью, несмотря на эпилепсию. Автор – известный детский эпилептолог профессор Ури Крамер. Издательство: клиника «Мигдаль Медикал» (Израиль, 2022)
Книга написана простым языком для мам и пап, полна практических советов и рекомендаций эксперта-эпилептолога с мировым именем.
В книге профессора Крамера вы найдёте ответы на многие ваши вопросы об эпилепсии у детей, начиная с видов приступов, правильной диагностики, эффективных методов лечения, и заканчивая практическими советами о том, как повысить качество жизни вашего ребёнка и подготовить его к самостоятельной взрослой жизни.
Синдром Веста у детей – симптомы и лечение приступов
Синдром Веста — это эпилептоподобное заболевание, характеризующееся развитием спастических мышечных сокращений на фоне отставания в нейропсихическом развитии и специфическими результатами электроэнцефалографии. Выявляется у детей в раннем возрасте. Для постановки диагноза проводятся ЭЭГ, компьютерная или магнитно-резонансная томография. Терапия основывается на применении противоэпилептических средств и стероидных гормонов. Возможно хирургическое лечение.
Общая информация

Синдром Веста: симптомы и лечение
Заболевание носит вторичный характер, так как в 90% случаев развивается на фоне органического поражения структур головного мозга. У небольшого числа больных установить причину не удается.
На патологию приходится 2-3% от всех случаев детской эпилепсии и около 25% от младенческой. Средняя распространенность среди новорожденных — 3-5 больных на 10 тыс. детей. Болезнь в 60% случаев встречается у мальчиков. Чаще всего первые признаки возникают до года. Пик манифестации — 4-6 месяц жизни. Картина заболевания меняется при взрослении. К возрасту 3 лет клинические проявления проходят, либо у пациента развивается эпилепсия другой формы.
Причины развития
Синдром Веста развивается на фоне первичного поражения головного мозга: энцефалит, гипоксические повреждения нервной системы, внутричерепная родовая травма, асфиксия новорожденного, внутриутробные инфекции и пр. Симптомы болезни отмечаются у новорожденных с аномалиями строения ЦНС: увеличением больших полушарий, недоразвитием мозолистого тела и т.п. Возможно появление эпилептических приступов на фоне факоматозов — нейрофиброматоза и туберозного склероза, и генетических болезней (инфантильные спазмы при фенилкетонурии, синдроме Дауна).
У 10% больных развитие патологии имеет идиопатический характер. Конкретная причина не может быть выявлена. Врачи считают, что в этих случаях болезнь носит наследственный характер и связана с генетическими аномалиями.
Механизм развития инфантильных спазмов продолжает изучаться. Основная гипотеза — нарушение работы нейронов, вырабатывающих серотонин. Это приводит к дисбалансу нейромедиаторов и может провоцировать развитие судорожных приступов. Характер подобных нарушений не объяснен. Другая теория связывает возникновение синдрома Веста с иммунологическими изменениями в организме, а именно с увеличением количества активных В-лимфоцитов. Гипотеза согласуется с эффективностью применения иммуносупрессивных препаратов в лечении.
Самолечение и использование народных методов терапии при судорожных синдромах недопустимо. Это может стать причиной прогрессирования патологии и развития осложнений.
Клинические проявления
Признаки заболевания появляются в первый год жизни. В редких случаях возможна манифестация патологии до 4-летнего возраста. Основные симптомы — отставание в психомоторном развитии и мышечные спазмы.
У большинства детей сначала возникают признаки нейропсихического развития, на фоне которых появляются судорожные приступы. У 30% больных мышечные спазмы являются первыми проявлениями. Родители отмечают, что у детей не развиты хватательный рефлекс, слежение за предметами, а также способность фиксировать свой взгляд. При внешнем осмотре заметна общая мышечная гипотония.
Спазмы мышц возникают внезапно. Они проявляются симметрично и проходят в течение 1 минуты. Для синдрома Веста характерно их серийное возникновение. Интервал между отдельными сокращениями различен. Количество приступов в сутки — от 1 до нескольких сотен. Чаще всего мышечные спазмы возникают утром после пробуждения или в вечернее время перед засыпанием. Громкий звук и тактильные прикосновения могут провоцировать спастические приступы.
Мышечные спазмы могут носить изолированный или генерализованный характер. В первом случае сокращения наблюдаются в определенной группе мышц. Их разделяют на несколько видов:
- разгибательные (экстензорные);
- сгибательные (флексорные);
- смешанные.
Смешанные мышечные спазмы встречаются наиболее часто. Реже наблюдаются изолированные сгибательные или разгибательные сокращения. Характер приступа определяется положением тела в момент его начала.
Локальные спазмы встречаются чаще генерализованных. Ребенок может кивать головой при поражении шейных мышц, или пожимать плечами. Характерный признак болезни — пароксизм «складного ножа», возникающий в результате спазма мышц брюшного пресса. Характерны сокращения мышц верхних конечностей. Больной при этом обхватывает свое туловище руками. У детей, которые умеют ходить, возникают дроп-атаки — внезапное падение при сохранении сознания.
Бессудорожные приступы встречаются редко. Они протекают по типу абсанса — ребенок замирает на одном месте на несколько секунд, после чего возвращается к прерванной деятельности. Инфантильные спазмы могут затрагивать только мышцы глазных яблок. Клиническая картина заболевания у всех детей имеет свои особенности.
Диагностические мероприятия

Обследование у детей носит комплексный характер
Триада признаков при синдроме Веста:
- гипсаритмический ЭЭГ-паттерн;
- отставание в психомоторном развитии;
- локальные мышечные спазмы.
В диагностике важную роль играет ранний возраст больного, что позволяет отличать заболевание от синдрома Драве и других болезней с судорожными проявлениями. Если болезнь возникает в возрасте 3-4 лет, то постановка диагноза затруднена. В процессе обследования ребенка осматривают педиатр, генетик, детский невролог и эпилептолог — они проводят дифференциальную диагностику.
Электроэнцефалография — «золотой стандарт» диагностики. Приступ на ЭЭГ имеет вид генерализованных медленноволновых комплексов с высокой амплитудой. Для них характерны периоды угнетения активности на 1 и более секунд. В межприступный период на ЭЭГ выявляется дезорганизованная беспорядочная спайк-волновая активность — гипсаритмия. Она характерна для периода сна и бодрствования.
Всем детям проводят компьютерную томографию головного мозга. Так как синдром Веста носит вторичный характер, то при нейровизуализации определяются очаговые и диффузные очаги поражения структур центральной нервной системы. Дополнительно возможно проведение МРТ или позитронно-эмиссионной томографии головного мозга.
Подходы к лечению
Длительное время заболевание было неизлечимо. В конце 60-х годов прошлого века был показан положительный эффект от приема препаратов адренокортикотропного гормона. Его применение и использование глюкокортикостероидов позволяет сократить число или полностью избавиться от спастических приступов, а также нормализовать картину мозговой активности на ЭЭГ. Схема лечения и дозировка лекарственных средств подбирается индивидуально. Средняя продолжительность терапии — 3-6 недель.
При выявлении первичного туберозного склероза назначаются медикаменты с вигабатрином. Это вещество обладающее сильным противоэпилептическим эффектом. Лечение им быстро приводит к снижению частоты спазмов. Среди других антиконвульсантов возможно назначение препаратов на основе вальпроевой кислоты. Положительный эффект имеют большие дозы витамина В6 при их введении на начальной стадии развития патологии.
Если консервативная терапия не дает эффекта, то проводятся нейрохирургические вмешательства. Конкретный вид операции определяет нейрохирург после осмотра ребенка.
Лекарственные средства имеют показания и противопоказания к применению. Их назначает только лечащий врач.
Прогноз
У 40-50% больных заболевание проходит при достижении 3-летнего возраста. В оставшихся случаях патология переходит в другой тип эпилептической патологии, например в синдром Леннокса-Гасто. В случае одновременного наличия синдрома Дауна болезнь плохо поддается консервативной терапии. Родителям важно понимать, что несмотря на устранение мышечных спазмов, задержка в психомоторном развитии сохраняется. Это приводит к поведенческим, когнитивным дефектам, а также к трудностям в обучении.
Говоря о том, сколько живут больные, врачи основывают прогноз на основании сопутствующих заболеваний. При наличии дефектов в строении головного мозга летальность достигает 25-30%. В других случаях уровень смертности не превышает 1%.
Синдром Веста – заболевание, имеющее благоприятный прогноз. Своевременное выявление болезни и комплексная терапия позволяют избавиться от спастических симптомов. Отставание в психомоторном развитии при этом сохраняется и требует длительной работы с ребенком для улучшения его адаптации. Диагностикой и терапией занимается врач-педиатр совместно с другими специалистами: неврологом и эпилептологом.
Видео


* Представленная информация не может быть использована для самостоятельной постановки диагноза, определения лечения и не заменяет обращение к врачу!
Синдром Веста : причины, симптомы, диагностика, лечение

Заболевания
Синдром Веста
Это тяжелая форма эпилепсии протекающие на фоне задержки нейропсихического развития и сопровождающиеся гипсаритмическим ЭЭГ-паттерном. Манифестирует в возрасте до 4-х лет, преимущественно на 1-ом году жизни. С момента первого клинического описания синдрома доктором Уильямом Вестом в 1841 году (на примере собственного сына) сформировалась классическая триада симптомов: эпилептические (инфантильные) спазмы; гипсаритмия на ЭЭГ и задержка или регресс развития.
Таким образом, СВ относится к группе «эпилептических энцефалопатий», которые характеризуются тяжелыми, лекарственно-устойчивыми эпилептическими приступами с началом в раннем возрасте (до 18 мес +-), патологическими изменениями на ЭЭГ и когнитивным дефицитом.
Что значит эпилептическая энцефалопатия простыми словами?
Не столько первопричина эпилепсии, сколько сами судороги или эпиактивность наносят основной вред развитию. В большинстве случаев, началу спазмов может предшествовать неврологическая симптоматика: потеря/неустойчивость зрительного контакта, гипотония, сонливость, аномальные рефлексы. Задержка развития варьирует от легкой до тяжёлой степени и появляется перед, с дебютом спазмов или спустя какое-то время после.
Так все ли пациенты с диагнозом синдром Веста будут с плохим исходом? – Нет.
Результаты ретроспективного исследования уровня интеллекта во взрослом возрасте на выборке из 147 пациентов с историей эпилептических спазмов (Riikonen R., 2020) показали:
* 25 детей ходили в обычную школу с IQ > 85;
* 11 пациентов имели незначительные нарушения с IQ от 68 до 85;
* 36 пациентов наблюдались с трудностью обучения лёгкой степени и IQ от 40 до 60;
* 75 с тяжелым нарушением с IQ < 40.
Также имеется значимая связь СВ и расстройства аутистического спектра (РАС): процент встречаемости достигает 13-20%. Полной ремиссии в течение одного года достигают порядка 40% пациентов, при длительном наблюдении в течение 20–35 лет после ремиссии спазмов судороги были зарегистрированы в трети случаев, у трети пациентов были судороги с ежедневной или ежемесячной частотой, а в оставшейся трети приступы случались реже раза в месяц. Спазмы, в целом, склоны исчезать в течение 3-4 лет и нередко трансформироваться в синдром Леннокса-Гасто (~в 18% случаев). Летальный исход регистрируется в 13-23% случаев (кумулятивный риск достигает 31%). Вероятность летального исхода значимо снижается при раннем контроле (*читай ремиссии) спазмов.
В долгосрочных наблюдениях четверть пациентов (25%) умерли к 17,2 годам, и 50% пациентов – к 48,6 годам. Большинство умерших пациентов имели тетрапарез и испытывали ограничения в повседневной активности.
До сих пор эффективность лечения синдрома Веста (СВ) невысока и ограничена синдромологическим подходом в выборе терапии. Выявление первопричины эпилепсии не только позволяет лучше спрогнозировать вектор течения заболевания, но и перейти к следующей модели лечения: этиопатогенетической. Отдельного внимания заслуживает быстроразвивающаяся область генотип-ориентированной модели лечения. Целенаправленное генетическое лечение – это надежда на будущее для тяжелых эпилепсий. Тем не менее, в современных реалиях, наша задача состоит в быстром выявлении СВ, так как скорость начала надлежащего лечения залог лучшего прогноза.
Классические проявления СВ:
– короткие (до 2 сек) эпизоды резкого сгибания туловища;
– с напряжением мускулатуры аксиальной (пресс) и плечевого пояса (руки);
– нередко организованы в кластеры или серии (идут друг за другом);
Запись ЭЭГ обычно показывает хаотичные высокоамплитудные медленные волны, на которые наслаиваются острые (разряды). Такая ЭЭГ картина носит название – гипсаритмия.
Психомоторная задержка или регресс в развитии связаны как с самими приступами, так и с патологической ЭЭГ, поэтому в случае с СВ мы действуем по принципу “всё или ничего”, что оправдывает достаточно агрессивную медикаментозную тактику.
«Единичные эпилептические спазмы (ЭС)» определяются как отсутствие других спазмов, возникающих в течение 1 мин до и после каждого ЭС. Соответственно при частоте эпизода раз в минуту, мы в праве говорить о серии.
Нюансы феноменологии (или как могут выглядеть ЭС):
– Спазмы могут проявиться в виде сгибания или разгибания.
– Могут сопровождаться эпизодами плача или
крика, которые могут предшествовать самому спазму или следовать за ним. Заболевшие дети после кризиса могут быть раздражительный или сонливы.
– В части фенотипов также могут наблюдаться отведения глаз, остановка дыхания, вегетативные проявления, гримасы и мигание/расширение глазных щелей.
На исход СВ влияют несколько факторов: ранний старт терапии, ответ на лечение, эволюция в сторону других эпилептических синдромов, степень задержки/регресса развития, общеклинические нарушения, вторичное ухудшение из-за побочных эффектов лечения. Рецидив приступов считается негативным фактором плохого прогноза, приводящим к более тяжёлой степени когнитивного дефицита. Наиболее значимыми прогностическими факторами являются этиология, возраст на момент дебюта, а также позднее и/или ненадлежащее лечение (об этом в следующем посте). Раннее и надлежащее лечение благоприятно влияет и на возможности долгосрочного развития, и на вероятность достижения ремиссии. Результат обычно плохой в случаях СВ, связанного с тяжелым поражением головного мозга (5-кратный риск летального исхода), пороками развития, постинфекционными заболеваниями или (определенными) генетическими причинами.

