
К вопросу о сложности диагностики синдрома Гудпасчера
Обсуждаются проблемы диагностики и лечения анти-БМК-ассоциированной болезни (синдрома Гудпасчера) – редкого, тяжелого прогрессирующего заболевания, ассоциированного с образованием антител к гломерулярной мембране [анти-БМК (anti-GBM)], при котором развивается легочно-почечный синдром, т.е. одновременное диффузное геморрагическое поражение легких в сочетании с острым или быстропрогрессирующим гломерулонефритом. Главная проблема – это поздняя диагностика заболевания, когда терапия заболевания уже малоэффективна. В статье приведены современные сведения об эпидемиологии, этиологии и патогенезе, принципах диагностики и лечения этого редкого заболевания, а также клиническое наблюдение пациента с синдромом Гудпасчера. Определены особенности течения болезни у данного больного.
Ключевые слова
Полный текст
АД – артериальное давление БМК – базальная мембрана клубочков СГ – синдром Гудпасчера СКФ – скорость клубочковой фильтрации СМП – скорая медицинская помощь СОЭ – скорость оседания эритроцитов ЧСС – частота сердечных сокращений ЭКГ – электрокардиография Системные васкулиты относительно редкие заболевания, которые имеют большое разнообразие клинических проявлений, являются сложными для диагностики и требуют проведения дифференциального диагноза с широким спектром патологических состояний. Клинические особенности той или иной формы системных васкулитов определяются патогенетическими механизмами развития. Одним из механизмов является поражение сосудов органоспецифическими антителами, что и наблюдается при развитии анти-БМК-ассоциированной болезни, известной как синдром Гудпасчера. Синдром Гудпасчера (СГ) – заболевание, ассоциированное с образованием антител к гломерулярной мембране [анти-БМК (anti-GBM)], при котором развивается легочно-почечный синдром, включающий в себя диффузное геморрагическое поражение легких в сочетании с острым или быстропрогрессирующим гломерулонефритом. Впервые термин «легочно-почечный синдром» использовал гарвардский патолог W. Goodpasture в 1919 г., описывая случай кровохарканья и гломерулонефрита у 18-летнего юноши, заболевшего после перенесенной инфлюэнцы. В 1958 г. М. Stanton и J. Tange описали 9 случаев сочетанного поражения легких и почек, сопровождавшихся рецидивирующими легочными кровотечениями, гемосидерозом легких и гломерулонефритом, с летальным исходом через несколько месяцев от начала заболевания. Они предложили назвать это редко встречающееся заболевание «синдром Гудпасчера» [1, 2]. СГ является редкой патологией, заболеваемость у взрослых составляет 0,5-1,0 на 1 000 000 в год, у детей значительно меньше. Выделяют два пика заболеваемости: в 20-30 и 60-70 лет. Мужчины болеют чаще женщин: соотношение частоты заболевания составляет 6-8:1 [1, 3, 4]. Причины, приводящие к развитию СГ, на сегодняшний день не установлены. Существуют данные о генетической предрасположенности к развитию заболевания. В качестве факторов, определяющих генетическую предрасположенность к СГ, рассматривают изменения в системе гистосовместимости. Есть предположение, что СГ ассоциирован с HLA-DR15, DRB1*1501, DRB1(*)1502 [4, 5]. К пусковым факторам у лиц с генетической предрасположенностью относят курение, вдыхание паров углеводородов и органических растворителей, воздействие инфекционных агентов (вирус гриппа, вирус иммунодефицита человека), ингаляции кокаина, металлической пыли, лекарственные препараты (D-пеницилламин, алемтузумаб) [1, 6]. Ключевым фактором развития СГ является образование антител к базальной мембране клубочков почек (анти-БМК), которые являются иммуноглобулинами IgG [1, 3, 7]. Базальная мембрана представляет собой многосоставную сложную структуру, разделяющую соединительную ткань, эндотелий или эпителий в организме. Одним из основных структурных компонентов мембраны служат нити коллагена IV типа, состоящие из α-цепей шести типов. Мишенью для анти-БМК являются два эпитопа неколлагенового домена α3-цепи нити коллагена IV типа, названные ЕА и ЕВ, а также у меньшего количества пациентов – эпитопы α5- и α4-цепей коллагена IV типа. Структура нитей коллагена не является уникальной только для клубочков почек или легочных альвеол, однако у пациентов с СГ основные патологические изменения развиваются преимущественно в легких в виде разрушения альвеол и развития синдрома диффузного альвеолярного кровотечения и в почках с развитием пролиферативного экстракапиллярного (быстропрогрессирующего) гломерулонефрита. Большое значение в повреждении органов также имеют активация системы комплемента и Т-лимфоциты, распознающие неколлагеновый домен α3-цепи коллагена IV типа [1, 3, 8]. Типичная клиническая картина заболевания, т.е. классический легочно-почечный синдром, с одновременным поражением легких в сочетании с гломерулонефритом наблюдается у подавляющего большинства пациентов – в 60-80% описанных случаев СГ. У части пациентов, примерно в 20%, имеется только поражение почек, реже встречаются только легочные проявления. Клинические изменения в легких при СГ могут проявляться как кровохарканьем, так и развитием альвеолярного кровотечения, приводить к прогрессирующей дыхательной недостаточности и гибели больного. Патологические изменения в почках приводят к развитию нефритического синдрома и формированию почечной недостаточности. Наряду с указанными синдромами у пациентов возможно развитие анемии, чаще железодефицитной, разной степени выраженности, появление болей в грудной клетке, гепатоспленомегалии, лихорадки, артралгии, миалгии, редко – двусторонний ретинит и увеит. При обследовании пациентов можно выявить характерные клинические признаки: кашель, кровохарканье, признаки дыхательной недостаточности (одышка, цианоз, гипоксемия), при аускультации легких – крепитация, влажные мелкопузырчатые хрипы, олигурия. По лабораторным данным – анемия, повышение скорости оседания эритроцитов (СОЭ), гематурия, протеинурия, повышение уровня сывороточного креатинина [1, 4, 7, 9]. При рентгенологическом исследовании – патогномоничны изменения в виде двусторонних узелковых и интерстициальных затемнений по типу «матового стекла» и консолидации легочной ткани [10]. Для подтверждения СГ наряду с выявлением у пациента легочно-почечного синдрома необходимо проведение серологического исследования крови и/или гистологического исследования почек или легких. Диагностическим признаком, позволяющим верифицировать СГ, является выявление анти-БМК методом иммуноферментного или радиоиммунного анализа (чувствительность и специфичность приближаются к 100%). Однако в единичных случаях анти-БМК в крови при СГ не выявляются, причины этого до конца не ясны, а также есть данные, указывающие на выявление анти-БМК у здоровых людей. Примерно у 1/3 пациентов с СГ наряду с высоким титром анти-БМК в крови определяют ANCA (антитела к миелопероксидазе, антитела к протеиназе-3), что не влияет на течение и прогноз заболевания [4]. При исследовании биоптатов почек обнаруживают анти-БМК в клубочках почек и гистологическую картину пролиферативного экстракапиллярного или некротизирующего гломерулонефрита. При биопсии легких, которая редко проводится, выявляется картина разрушенных альвеол, альвеолярные пространства заполнены эритроцитами, скоплениями макрофагов, нагруженных гемосидерином [2, 4, 8, 9]. СГ необходимо дифференцировать с рядом заболеваний, протекающих с похожей клинической картиной: острым гломерулонефритом (ANCA-ассоциированный некротизирующий фокальный и сегментарный гломерулонефрит), тяжелой пневмонией, ANCA-ассоциированными васкулитами [гранулематоз с полиангиитом (гранулематоз Вегенера), микроскопический полиангиит, синдром Черджа-Стросса], с системной красной волчанкой. Основными методами лечения СГ являются применение плазмафереза с целью элиминации анти-БМК из крови, а для предотвращения их дальнейшего образования и подавления тканевого воспаления – иммуносупрессивная терапия: преднизолон, циклофосфамид, возможно использование ритуксимаба, применение которого, по данным литературы, приводит к значительным улучшениям состояния. При выраженном синдроме альвеолярного кровоизлияния и дыхательной недостаточности необходима искусственная вентиляция легких (ИВЛ), при формировании тяжелой почечной недостаточности – проведение гемодиализа и, возможно, трансплантации почки. При отсутствии адекватной терапии СГ имеет злокачественное течение, приводящее к быстрой гибели пациентов от прогрессирующей дыхательной недостаточности, легочного кровотечения, почечной недостаточности [1, 9]. Сообщения о каждом новом случае СГ в настоящее время продолжают представлять большой научный интерес в связи с тем, что СГ относительно редко встречающееся заболевание, и еще многое остается неясным в его этиологии и патогенезе. В нашей клинике за последние 25 лет нам довелось наблюдать 3 больных с СГ. В качестве примера приводим последнее клиническое наблюдение пациента со злокачественно протекающим СГ. Больная К., 56 лет, находилась на стационарном лечении в терапевтической клинике УКБ №4 ПМГМУ им. И.М. Сеченова в марте 2016 г. Пациентка доставлена в клинику скорой медицинской помощью (СМП) с направительным диагнозом: Внебольничная двусторонняя полисегментарная пневмония тяжелого течения. Дыхательная недостаточность II степени. При поступлении в стационар в связи с тяжестью состояния сразу из приемного отделения госпитализирована в отделение реанимации и интенсивной терапии. Пациентка предъявляла жалобы на кашель с выделением крови алого цвета, одышку в покое, повышение температуры тела до 38,2°С, головокружение, выраженную общую слабость. В анамнезе вредные привычки отсутствуют, воздействию профессиональных вредностей не подвергалась. Операции не проводились. Аллергологический и семейный анамнез не отягощены. С детства наблюдалась по поводу эпилепсии, принимала карбамазепин. В течение 5 лет имеется повышенное артериальное давление (АД) до 180/90 мм рт. ст., в качестве антигипертензивной терапии постоянно принимала лозартан 50 мг в сутки и метопролол 50 мг в сутки. При сборе анамнеза установлено, что ухудшение состояния наступило с осени 2015 г., когда больная отметила снижение аппетита, появление общей слабости, уменьшение массы тела – в феврале 2016 г. похудела на 18 кг. Также в октябре 2015 г. появился дискомфорт в поясничной области и неоднократно эпизоды покраснения мочи. С указанными выше жалобами в феврале 2016 г. больная госпитализирована в нефрологическое отделение городской больницы г. Москвы, где поставлен диагноз «Нефропатия смешанного генеза за счет лекарственного интерстициального нефрита и гипертонического ангиосклероза с развитием хронической болезни почек Ⅳ стадии [скорость клубочковой фильтрации (СКФ) 16 мл/мин]», иммуносупрессивная терапия не проводилась. После окончания стационарного лечения больной рекомендовано наблюдение нефрологом по месту жительства. На следующий день после выписки из нефрологического отделения у больной повысилась температура тела до 38,2°С, появился кашель с выделением крови в большом количестве, одышка, которая быстро прогрессировала, в связи с чем пациентка вызвала СМП и была экстренно госпитализирована в нашу клинику. При физическом обследовании общее состояние больной тяжелое, сознание ясное. Телосложение правильное, конституция нормостеническая, рост 155 см, масса тела 89 кг, индекс массы тела 37 кг/м2. Кожные покровы бледные, акроцианоз, цианоз губ, лимфатические узлы, доступные для пальпации, не увеличены, безболезненны. Кости, мышцы, суставы без видимых изменений. Со стороны органов дыхания: тахипноэ – частота дыхательных движений до 28 в минуту, снижение сатурации крови до 89%, свидетельствующие о дыхательной недостаточности; при перкуссии – притупление перкуторного звука в нижних и средних отделах грудной клетки с обеих сторон, при аускультации – влажные мелкопузырчатые хрипы по всем легочным полям. При кашле больная выделяла большое количество мокроты алого цвета, объем которой соответствовал легочному кровотечению. Область сердца и крупных сосудов визуально не изменена, границы сердца смещены влево (левая граница на 1 см кнаружи от левой среднеключичной линии), тоны сердца приглушены, частота сердечных сокращений (ЧСС) 96 ударов в минуту, ритм правильный, АД 160/100 мм рт. ст. Со стороны органов пищеварения патологии не выявлено, живот при пальпации мягкий, безболезненный, размеры печени по Курлову 9×8×8 см. Селезенка не увеличена, не пальпируется. Стул регулярный. Область почек визуально не изменена, почки не пальпируются. Неврологический статус: сознание ясное, в пространстве и времени ориентирована правильно. Клинический анализ крови: выявлена гипохромная анемия – снижение гемоглобина до 71 г/л (в динамике – снижение до 51 г/л), лейкоцитоз 12,6×109/л, увеличение СОЭ до 36 мм/ч. При биохимическом исследовании крови при поступлении: гипопротеинемия – общий белок 43 г/л, повышение уровня мочевины и креатинина до 15,3 ммоль/л и 520 мкмоль/л соответственно, калий 4,6 ммоль/л, железо 3,2 мкмоль/л. СКФ, рассчитанная по CKD-EPI, 7,4 мл/мин. В повторном анализе через 7 дней: уровень мочевины – 45,1 ммоль/л, креатинина – 451 мкмоль/л, калия – 5,75 ммоль/л, СКФ – 8,7 мл/мин. При исследовании показателей свертывающей системы крови выявлено снижение уровня протромбинового индекса до 71%, повышение уровня D-димера до 4,1 мг/л. В общем анализе мочи: гематурия – эритроциты покрывают все поле зрения, белок – 0,21 г/л, лейкоциты – единичные. Анализ мочи по Нечипоренко: лейкоциты – 3000 в 1 мл, эритроциты – 562 500 в 1 мл, цилиндры – 250 в 1 мл. Электрокардиография (ЭКГ; 18.02.16): ритм правильный, горизонтальное положение электрической оси сердца, синусовая тахикардия, ЧСС 92 в 1 мин; в динамике по ЭКГ сохранялся синусовый ритм, горизонтальное положение ЭОС. Эхокардиография (20.02.16): признаки гипертрофии левого желудочка. Ультразвуковое исследование органов брюшной полости и забрюшинного пространства (19.02.16): выявлена повышенная эхогенность печени и поджелудочной железы, без изменения контуров и размеров этих органов, размеры почек были нормальными (правая – 100×37 мм, левая – 101×38 мм), контуры почек неровные, размер паренхимы 9–16 мм с обеих сторон, эхогенность паренхимы повышена, синусы почек уплотнены, расширение чашечно-лоханочной системы и конкременты не выявлены. Селезенка без патологии. Эзофагогастродуоденоскопия (19.02.16): признаки поверхностного гастрита, недостаточность кардии. Рентгенография органов грудной клетки (18.02.16): инфильтраты по всем легочным полям. Мультиспиральная компьютерная томография органов грудной клетки (19.02.16): по всем сегментам легких распространенные участки уплотнения легочной ткани по типу «матового стекла» и консолидации, с прослеживаемой на фоне данных изменений воздушной бронхограммой. В динамике отмечалось увеличение протяженности участков инфильтрации легочной ткани с обеих сторон, понижение пневматизации легочной ткани в нижней доле справа с переходом зон «матового стекла» в консолидацию легочной ткани (рис. 1). Видеобронхоскопия (19.02.16): эндоскопическая картина состоявшегося легочного кровотечения, хронического трахеобронхита на фоне очаговой атрофии слизистой оболочки. Учитывая наличие клинической картины легочно-почечного синдрома, заподозрен СГ и проведено иммунологическое исследование, при котором выявлены антитела к миелопероксидазе (cANCA) – 40,8 ЕД/мл (норма 0-5 ЕД/мл), антитела к протеиназе (pANCA) – 0,27 ЕД/мл (норма 0-5 ЕД/мл), а также анти-БМК – 22 ЕД/мл (норма – до 20 ЕД/мл). Биопсия почки не выполнялась по причине крайне тяжелого состояния пациентки. Таким образом, на основании клинических данных и результатов иммунологического исследования, положительного анализа на анти-БМК, больной поставлен диагноз: Синдром Гудпасчера. Больной проводилась следующая терапия: метилпреднизолон в дозе 1000 мг внутривенно, циклофосфан 600 мг в сутки внутривенно. Проводимая медикаментозная терапия была не эффективна, у больной нарастала дыхательная недостаточность, которая потребовала проведения ИВЛ с третьего дня пребывания в клинике. Состояние пациентки прогрессивно ухудшалось – тяжесть состояния обусловлена тяжелой дыхательной и почечной недостаточностью. На девятый день с момента поступления (27.02.16) у больной развилась брадикардия 28 в минуту, гипотония 50/20 мм рт. ст., на кардиомониторе – идеовентрикулярный ритм с переходом в асистолию. Несмотря на реанимационные мероприятия, наступила биологическая смерть больной. Проведено патологоанатомическое исследование, выявлены изменения со стороны легких и почек, характерные для СГ и подтвердившие клинический диагноз. Легкие с неоднородной консистенцией, пестрого вида: очаги белесоватого цвета, размерами до 1×2×1 см, возвышающиеся над поверхностью разреза, контрастировали с обширными сливающимися участками красновато-буроватого цвета. Почки увеличены в размерах, дрябловатой консистенции, корковый слой расширен, тусклый, желтовато-сероватый с мелким красным крапом. При гистологическом исследовании легких выявлено утолщение межальвеолярных перегородок с инфильтрацией эозинофилами и нейтрофилами, пролиферация альвеолоцитов. В просвете альвеол – большое число эритроцитов, сидероцитов. В почечных клубочках отмечены полулуния, образованные в одних клубочках клеточными элементами, в других – фиброзной тканью (см. рис. 2 на цветной вклейке). Таким образом, данные аутопсии подтвердили клинический диагноз. Заключение Особенностью данного клинического случая является развитие СГ у женщины, чаще заболевание развивается у лиц мужского пола. СГ дебютировал в виде гломерулонефрита с последующим, спустя 5 мес, присоединением поражения легких в виде выраженного быстропрогрессирующего синдрома альвеолярного кровоизлияния с развитием тяжелой дыхательной недостаточности, что привело к летальному исходу. Авторы заявляют об отсутствии конфликта интересов.
Синдром Гудпасчера : симптомы, лечение, клинические рекомендации
Болезнь Гудпасчера характеризуется легочными кровотечениями и гломерулонефритом. Заболевание связано с выработкой антител, часто направленных против отдельных эпитопов коллагена IV типа в базальной мембране легочных альвеол и почечных клубочков. Причина неизвестна. Болезнь Гудпасчера следует отличать от синдрома Гудпасчера, при котором легочное кровотечение и гломерулонефрит обусловлены различными системными нарушениями, включая СКВ, геморрагический васкулит, узелковый полиартериит и гранулематоз Вегенера. У некоторых больных нефрит с антителами к базальной мембране почечных клубочков развивается в отсутствие легочного кровотечения и представляет собой одну из форм быстропрогрессирующего гломерулонефрита.
Патоморфология болезни Гудпасчера. Изменения при световой микроскопии в большинстве случаев напоминают быстропрогрессирующий гломерулонефрит. Иммунофлюоресцентная микроскопия выявляет линейные отложения IgG по ходу базальной мембраны клубочков.
Клинические проявления болезни Гудпасчера. Болезнь Гудпасчера редко встречается в детском возрасте. Обычно возникает кровохарканье, обусловленное легочным кровотечением, которое может быть опасным для жизни. Поражение почек проявляется острым нефритическим синдромом с гематурией, протеинурией и повышением АД и прогрессирует за несколько дней или недель. Уровень компонента комплемента С3 в сыворотке крови остается нормальным.
Диагностика болезни Гудпасчера. Биопсия почек позволяет предположить болезнь Гудпасчера. Присутствие в сыворотке антител к базальной мембране почечных клубочков подтверждает диагноз и исключает другие заболевания, сопровождающиеся синдромом Гудпасчера.
Прогноз и лечение болезни Гудпасчера. У больных, выживших после легочного кровотечения, обычно развивается терминальная стадия ХПН. Пульс-терапия метилпреднизолоном в сочетании с циклофосфамида и плазмаферезом увеличивает выживаемость и способствует устранению почечной патологии.
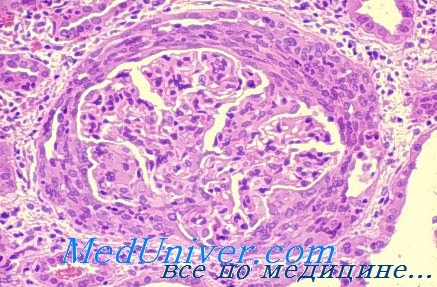
Гистология почек при болезни Гудпасчера
Гемолитико-уремический синдром
Гемолитико-уремический синдром (ГУС) — самая частая причина острой почечной недостаточности у маленьких детей, характеризуется микроангиопатической гемолитической анемией, тромбоцитопенией и уремией. Этот синдром сходен с тромботической тромбоцитопенической пурпурой, однако последняя обычно наблюдается у молодых женщин, рецидивирует, сопровождаясь лихорадкой, тяжелыми нарушениями ЦНС и тромбоцитопенией.
В развивающихся странах гемолитико-уремический синдром (ГУС) в 80 % случаев предшествует острый энтерит с поносом, вызываемый штаммом Escherichia coli (серотип 0157:Н1), который продуцирует веротоксин, близкий к цитотоксину Shigella dysenteriae. Этот микроб содержится в ЖКТ домашнего скота и попадает в организм человека с недожаренным мясом и непастеризованным молоком. Наблюдались вспышки заболевания после потреблении загрязненных напитков или купания в загрязненной воде.
Патоморфология гемолитико-уремического синдрома. Начальные изменения клубочков сводятся к утолщению стенок капилляров, сужению их просвета и разрастанию мезангия. При электронной микроскопии видно, что эти изменения обусловлены гранулярным отложением аморфного вещества неизвестного происхождения в субэндотелиальном слое и между капиллярами. В самих капиллярах и артериолах обнаруживаются фибриновые тромбы, которые могут приводить к некрозу коркового вещества почек.
В одних клубочках изменения прогрессируют вплоть до частичного или полного склероза, другие атрофируются вследствие ишемии. В пораженных мелких артериях и артериолах происходит концентрическая пролиферация интимы, закупоривая сосуды.
Патогенез гемолитико-уремического синдрома. Основную роль в патогенезе синдрома играет повреждение эндотелиальных клеток. Повреждение эндотелия почечных капилляров и артериол приводит к образованию отдельных тромбов. Диссеминированная внутрисосудистая коагуляция отмечается редко. Механическое повреждение эритроцитов при их прохождении через суженные участки сосуда обусловливает микроангиопатическую анемию. В основе тромбоцитопении лежат внутрипочечная адгезия или повреждение кровяных пластинок. Печень и селезенка удаляют поврежденные эритроциты и тромбоциты из кровотока. Эпизоды ГУС без диареи и отдельные семейные случаи могут объясняться отсутствием в плазме фактора, стимулирующего продукцию эндотелием простациклина, который расширяет сосуды и препятствует образованию тромбов.
Снижение уровня тромбомодулина, тканевого активатора плазминогена (алтеплазы) и гепариноподобных молекул, активирующих антитромбин III, напротив, способствует образованию тромбов при этом синдроме. Кроме того, еще до развития почечной патологии в сыворотке возрастает содержание таких прокоагулянтов, как фактор активации тромбоцитов, фрагменты протромбина 1 и 2, антиген тканевого активатора плазминогена, комплекс тканевого активатора и ингибитора плазминогена (ТАП-1), фактор Виллебранда, D-димеры и тромбоксан А2, которые и могут быть причиной тромбообразования.
Синдром Гудпасчера
Синдром Гудпасчера представляет собой иммуно-воспалительное заболевание мелких сосудов легких и почек.
Симптомы Синдрома Гудпасчера:
Классической триадой, выражающей клинико-патогенетическую сущность этой болезни, являются легочные кровотечения, гломерулонефрит и антитела к антигенам основной мембраны капилляров легких и почек. Заболевание встречается редко и может поражать любой возраст, но чаще болеют молодые мужчины. Конкретные причины неизвестны; не раз описывалось развитие синдрома Гудпасчера после недавно перенесенного гриппа или вдыхания углеводородов. В связи с этим не исключено, что подобные воздействия таким образом изменяют химическую структуру упомянутых выше антигенов основных мембран, что они приобретают аутоантигенные свойства и вызывают продукцию соответствующих антител с патогенными свойствами.
В тканях почек с помощью иммунофлюоресцентного метода обнаруживают линейные отложения антител к базальным мембранам, сочетающиеся в ряде случаев с отложениями фракции комплемента С3. В то же время при электронномикроскопическом исследовании не обнаруживают отложений иммунных комплексов. Циркулирующие антитела к базальным мембранам канальцев, клубочков и легочных альвеол в сыворотке крови обнаруживают более чем у 90% больных (особенно в ранней стадии заболевания), однако они не отражают тяжести органных изменений или общего прогноза, имея, таким образом, в основном диагностическое значение. Уровень сывороточного комплемента и циркулирующих иммунных комплексов в норме. При гистологическом исследовании почечные клубочки у некоторых больных могут выглядеть нормальными, но в большинстве случаев наблюдается явная патология – от очаговых пролиферативных изменений до некротического гломерулонефрита. Наиболее часто развиваются экстракапиллярные “полулуния”. В легких находят кровоизлияния в полость альвеол с картиной альвеолита или без нее.
В начале заболевания клинические проявления связаны прежде всего с поражением легких – кашель, небольшая одышка и особенно кровохарканье; реже наблюдается легкий цианоз. При аускультации возможны влажные хрипы. Рентгенологически характерны инфильтраты разной величины в обоих легких, особенно в прикорневых областях. В мокроте, как правило, обнаруживают макрофаги, содержащие железо. Повышенное отложение железа в легочной ткани может быть установлено сканированием с ^Ре. У многих больных имеют место лихорадка, боль в суставах, резкая общая слабость, но в ряде случаев эти симптомы выражены слабо или отсутствуют. У большинства больных с первых дней или недель заболевания регистрируют признаки гломерулонефрита. В анализах крови обнаруживают повышенную СОЭ и лейкоцитоз; позже в результате легочных кровотечений возможна гипохромная анемия.
Течение болезни в целом неблагоприятное, хотя и неоднотипное. На первых этапах основной угрозой являются легочные геморрагии, причем нередко больной умирает в результате первого и единственного профузного кровотечения. У ряда переживших это кровотечение может развиться относительная ремиссия легочного процесса, но во многих случаях кровохарканье и легочные геморрагии рецидивируют. Поражение почек у отдельных больных остается относительно нетяжелым, но в большинстве случаев быстро прогрессирует с развитием олигурической почечной недостаточности, приводящей к смерти в течение нескольких месяцев и даже недель. Средняя продолжительность жизни у неадекватно леченных больных составляет менее полугода.
Диагностика Синдрома Гудпасчера:
При дифференциальном диагнозе синдрома Гудпасчера следует иметь в виду, что сочетание легочного кровотечения с выраженной почечной патологией (в том числе с недостаточностью почек) может встретиться при гранулематозе Вегенера, узелковом полиартериите, СКВ, геморрагическом васкулите, криоглобулинемии, эмболии ветвей легочной артерии вследствие тромбоза почечной вены, болезни легионеров и даже при тяжелой недостаточности кровообращения с выраженным застоем в легких и почках. Однако все эти заболевания отличаются от синдрома Гудпасчера по клиническим признакам и отсутствию антител к антигенам базальных мембран.
Лечение Синдрома Гудпасчера:
Современная лекарственная терапия несколько улучшила прогноз синдрома Гудпасчера. При угрожающих легочных кровотечениях показано парентеральное введение метилпреднизолона по типу пульс-терапии (500-1000 мг в день), после чего переходят на длительный прием кортикостероидов внутрь (по 40- 80 мг преднизолона в сутки с очень медленным и постепенным снижением дозы). При отсутствии тяжелых кровотечений стероиды назначают внутрь с начала лечения. Имеются указания на нарастание эффекта в случаях их сочетания с длительным приемом иммунодепрессантов – азатиоприна или циклофосфамида по 150-200 мг в день. Значительное улучшение почечных изменений и практическая ремиссия легочного процесса описаны у больных, которым назначали повторные курсы плазмафереза (с обменом 2-4 л плазмы в день процедуры) в комбинации с рассмотренным выше лекарственным лечением. Положительный эффект наблюдался даже у больных с уже сформированными пролиферативными изменениями (“полулуниями”) в почечных клубочках. Следует помнить, что при легочных кровотечениях противопоказаны антикоагулянты, которые назначают при других васкулитах. У больных с почечной недостаточностью на фоне необратимых морфологических изменений (выраженный фиброз, облитерация клубочков, атрофия канальцев) рассчитывать на улучшение можно только в результате применения регулярного гемодиализа либо трансплантации почки (хотя в трансплантированной почке в последующем не исключается развитие гломерулонефрита). Обобщая, следует признать, что современная терапия привела к некоторому улучшению течения синдрома Гудпасчера, но не привела к принципиальным успехам. Тем не менее можно полагать, что заболевание в принципе обратимо, в пользу чего свидетельствуют единичные его спонтанные ремиссии.
К каким докторам следует обращаться если у Вас Синдром Гудпасчера:
Вас что-то беспокоит? Вы хотите узнать более детальную информацию о Синдрома Гудпасчера, ее причинах, симптомах, методах лечения и профилактики, ходе течения болезни и соблюдении диеты после нее? Или же Вам необходим осмотр? Вы можете записаться на прием к доктору .
Синдром Гудпасчера
Синдром Гудпасчера – аутоиммунная патология, характеризующаяся образованием аутоантител к базальным мембранам почечных клубочков и легочных альвеол. Клинически синдром Гудпасчера проявляется рецидивирующими легочными кровотечениями, прогрессирующим гломерулонефритом и почечной недостаточностью. Диагноз синдрома Гудпасчера подтверждается выявлением антител к базальной мембране клубочков (Anti GBM), данными биопсии почек и легких, рентгенологического исследования легких. Лечение синдрома Гудпасчера включает иммуносупрессивную терапию (глюкокортикостероиды, цитостатики), плазмаферез, по показаниям – гемодиализ, трансплантацию почек.
Общие сведения
Синдром Гудпасчера – иммуно-воспалительное поражение капилляров почек и легких, протекающее с развитием гломерулонефрита и геморрагического пневмонита. Впервые признаки данной патологии были описаны в 1919 г. американским патофизиологом Э.У. Гудпасчером, за что болезнь и была названа его именем. В ревматологии синдром Гудпасчера относится к системным васкулитам и нередко обозначается как «геморрагический легочно-почечный синдром», «геморрагическая пневмония с гломерулонефритом», «идиопатический гемосидероз легких с нефритом». Частота развития синдрома Гудпасчера составляет 1 случай на 1 млн. населения. Отмечается два возрастных пика заболеваемости – в 20-30 лет и 50-60 лет; болеют преимущественно мужчины. При отсутствии лечения синдрома Гудпасчера летальность среди пациентов достигает 75-90%.
Причины синдрома Гудпасчера
Этиологические механизмы заболевания достоверно не определены. Клинические наблюдения указывают на связь развития синдрома Гудпасчера с перенесенной вирусной инфекцией (гриппом, вирусным гепатитом А и др.), приемом лекарственных препаратов (карбимазола, пеницилламина), производственными вредностями (вдыханием паров органических растворителей, лаков, бензина), переохлаждением, курением. Отмечена генетическая предрасположенность к данному синдрому у лиц–носителей HLA-DRwl5, HLA-DR4 и HLA-DRB1 аллелей. Описаны семейные случаи синдрома Гудпасчера.
Под воздействием того или иного этиологического фактора, в результате изменения толерантности иммунной системы, в организме начинается выработка аутоантител к базальным мембранам легочных альвеол и почечных клубочков. Предполагается, что в роли аутоантигена выступает структурный компонент а-3 цепи коллагена IV типа, присутствующий в базальных мембранах легочных и почечных капилляров. Сформировавшиеся антитела (GВМ-антитела) в присутствии С3-комплемента связываются с антигенами; образовавшиеся иммунные комплексы откладываются вдоль базальных мембран, индуцируя иммуно-воспалительное поражение почечных клубочков (гломерулонефрит) и альвеол (альвеолит). В развитии аутоиммунного воспаления большая роль принадлежит активации клеточных элементов (Т-лимфоцитов, эндотелиоцитов, моноцитов, альвеолярных макрофагов, полиморфноядерных лейкоцитов), цитокинов (инсулиноподобного, тромбоцитарного факторов роста, факторов некроза опухоли, интерлейкина-1), свободных радикалов, протеолитических ферментов и других факторов, повреждающих почечную и легочную ткань.
Патоморфологическими субстратами синдрома Гудпасчера служат геморрагический некротизирующий альвеолит и нефрозонефрит. При гистологическом исследовании почечной ткани обнаруживается пролиферативно-мембранозный, пролиферативный или некротизирующий гломерулонефрит, склероз клубочков и фиброз почечной паренхимы. Морфологическое исследование легочной ткани выявляет капиллярит межальвеолярных перегородок, легочные инфильтраты, гемосидероз, пневмосклероз.
Симптомы синдрома Гудпасчера
Выделяют три варианта клинического течения синдрома Гудпасчера: злокачественный, умеренный и медленный. Для злокачественного течения характерны рецидивирующая геморрагическая пневмония и стремительно прогрессирующий гломерулонефрит. При втором типе легочно-почечный синдром развивается медленнее и выражен умеренно. При третьем варианте синдрома Гудпасчера преобладают явления гломерулонефрита и ХПН; легочные проявления развиваются поздно.
Злокачественный вариант синдрома Гудпасчера дебютирует легочным кровотечением и острой почечной недостаточностью, требующими проведения интенсивной терапии (устранения водно-электролитных нарушений, возмещения кровопотери, ингаляций кислорода, ИВЛ, гемодиализа или перитонеального диализа). В других случаях заболевание может начинаться с общих симптомов: субфебрилитета, недомогания, похудания. Иногда появлению жалоб предшествует перенесенная ОРВИ. Из специфических симптомов обычно первыми развиваются признаки поражения легких – кашель, прогрессирующая одышка, цианоз, боль в грудной клетке, рецидивирующее кровохарканье или легочное кровотечение. Поражение легких при синдроме Гудпасчера нередко осложняется сердечной астмой и отеком легких.
Вскоре к легочным проявлениям добавляется почечная симптоматика: гематурия, олигурия, периферические отеки, артериальная гипертензия. У 10-15% пациентов синдром Гудпасчера манифестирует с клинических признаков гломерулонефрита. Во многих случаях течение заболевания сопровождается миалгиями, артралгиями, геморрагиями кожи и слизистых оболочек, интраретинальными кровоизлияниями, перикардитами.
Диагностика синдрома Гудпасчера
При осмотре пациентов с синдромом Гудпасчера обращает на себя внимание бледность кожных покровов, пастозность или отеки лица. В легких выслушиваются сухие и влажные хрипы, количество которых увеличивается в момент кровохарканья и после него. В общем анализе крови обнаруживается гипохромная анемия, анизоцитоз, пойкилоцитоз, лейкоцитоз, резкое увеличение СОЭ. Для общего анализа мочи характерна протеинурия, цилиндрурия, эритроцитурия; проба по Зимницкому выявляет изогипостенурию. В биохимическом анализе крови определяется нарастание уровня креатинина, мочевины, серомукоида; снижение концентрации железа. Для синдрома Гудпасчера типично обнаружение большого количества эритроцитов, сидерофагов и гемосидерина в общем анализе мокроты.
Наиболее чувствительным и специфичным методом диагностики синдрома Гудпасчера служит определение антител к базальной мембране клубочков (Anti-GBM) с помощью ИФА или РИА. На рентгенограммах легких выявляются множественные очаговые тени. Морфологическое подтверждение синдрома Гудпасчера основывается на данных биопсии легких и почек. Вспомогательное значение имеют результаты инструментальной диагностики: спирометрии, УЗИ почек, ЭКГ, ЭхоКГ.
Лечение и прогноз синдрома Гудпасчера
В остром периоде синдрома Гудпасчера показано назначение пульс-терапии метилпреднизолоном или комбинированной пульс-терапии (метилпреднизолон+циклофосфан) с последующим переводом на поддерживающую терапию после достижения клинико-лабораторной и рентгенологической ремиссии. С целью элиминации циркулирующих иммунных комплексов проводится плазмаферез. Симптоматическая терапия синдрома Гудпасчера включает переливания эритроцитарной массы и плазмы крови, назначение препаратов железа. При развитии терминальной почечной недостаточности применяются сеансы гемодиализа. Возможно выполнение нефрэктомии с последующей трансплантацией почки, однако это не исключает рецидива некротизирующего гломерулонефрита в трансплантате.
Течение синдрома Гудпасчера неуклонно прогрессирующее; прогноз – мало обнадеживающий. Гибель пациентов происходит, как правило, вследствие профузных легочных кровотечений, тяжелой почечной или дыхательной недостаточности. При злокачественном варианте летальный исход наступает в считанные недели; в остальных случаях средняя продолжительность жизни колеблется от нескольких месяцев до 1-3 лет. В литературе описаны единичные спонтанные ремиссии синдрома Гудпасчера.
Публикации в СМИ
Синдром Гудпасчера — аутоиммунное заболевание, характеризующееся присутствием АТ к коллагену типа IV c a -3-цепью (Гудпасчера Аг). Указанный Аг входит в состав базальной мембраны почечных клубочков и альвеолярной мембраны, поэтому основными клиническими проявлениями синдрома Гудпасчера выступают лёгочное кровотечение и прогрессирующий гломерулонефрит. Статистические данные. Распространённость: 0,5 на 1 000 000. Возраст: 5–40 лет. Преобладающий пол — мужской (6:1).
Этиология неизвестна. К факторам риска относят: курение, респираторную инфекцию, контакт с летучими углеводородами.
Генетические аспекты. Выявлена ассоциация с HLA DRw2.
Патоморфология • Почки •• Эпителиальноклеточные полулуния •• Сморщивание почечных клубочков •• Интерстициальный воспалительный экссудат • Лёгкие •• Внутриальвеолярные кровоизлияния •• Макрофаги, нагруженные гемосидерином •• Фиброз межальвеолярных перегородок.
Клиническая картина • Поражение лёгких (опережает поражение почек на несколько недель или месяцев); варьирует от небольшой одышки до повторных лёгочных кровотечений с развитием дыхательной недостаточности • Быстропрогрессирующий гломерулонефрит с развитием почечной недостаточности • Артериальная гипертензия (20%) • Гриппоподобный синдром: лихорадка, миалгии, артралгии, слабость.
Лабораторные данные • ОАК •• гипохромная ЖДА • ОАМ •• микрогематурия •• протеинурия, не достигающая нефротического порога • АТ против Аг базальной мембраны клубочков почек (радиоиммунный метод или метод непрямой иммунофлюоресценции) • Перинуклеарные антинейтрофильные АТ.
Инструментальные данные • Биопсия почек — отложения иммуноглобулинов и комплемента в базальной мембране клубочков, экстракапиллярные полулуния в 50% клубочков • Рентгенография органов грудной клетки — летучие асимметричные облаковидные инфильтраты; деструкция лёгких не характерна.
Дифференциальная диагностика • Гранулематоз Вегенера протекает с деструкцией лёгочной ткани и поражением верхних дыхательных путей • Микроскопический полиангиит, помимо лёгочного и почечного синдромов, имеет и другие (множественный мононеврит, кожные изменения) • Тромбоз почечных вен и ТЭЛА (острое развитие, отсутствие иммунологической активности) • Быстропрогрессирующий гломерулонефрит, осложнённый отёком лёгких.
ЛЕЧЕНИЕ
Общая тактика предполагает сочетание плазмафереза, активной иммунодепрессивной терапии и методов коррекции почечной функции. Диета соответствует диете при почечной недостаточности.
Лекарственное лечение • ГК •• Пульс-терапия метилпреднизолоном 30 мг/кг/сут в/в капельно в течение 20–30 мин 3 дня подряд •• Преднизолон после пульс-терапии в дозе 2 мг/кг внутрь до клинического эффекта с постепенным снижением дозы: до 1,75 мг/кг в течение 1 мес, 1,5 мг/кг в течение 3 мес, далее каждую дозу назначают по 6 мес (1,25–1,0–0,75–0,5–0,25 мг/кг), далее каждую дозу назначают по 12 мес (0,125–0,0625 мг/кг). Больным старше 60 лет дозу следует снизить на 25%.
• Цитотоксические иммунодепрессанты применяют в комбинации с ГК •• Циклофосфамид 2–3 мг/кг/сут •• Азатиоприн 1–2мг/кг/сут. Примечания: дозу препарата следует уменьшить на 50% при снижении СКФ до 10 мл/мин; необходим контроль за количеством лейкоцитов и тромбоцитов крови.
Немедикаментозная терапия • Плазмаферез ежедневно или через день в сочетании с иммунодепрессивной терапии в течение 1–2 нед • Гемодиализ — при развитии почечной недостаточности.
Хирургическое лечение • Трансплантация почки в терминальной стадии ХПН. Рецидива заболевания в трансплантате не наблюдают, если перед операцией в крови не обнаруживались АТ к базальной мембране клубочков.
Прогноз неблагоприятен, если на момент постановки диагноза (до начала лечения) уже имеются признаки почечной недостаточности.
Синоним. Синдром лёгочно-почечный наследственный.
МКБ-10 • M31.0 Гиперчувствительный ангиит
Код вставки на сайт
Синдром Гудпасчера
Синдром Гудпасчера — аутоиммунное заболевание, характеризующееся присутствием АТ к коллагену типа IV c a -3-цепью (Гудпасчера Аг). Указанный Аг входит в состав базальной мембраны почечных клубочков и альвеолярной мембраны, поэтому основными клиническими проявлениями синдрома Гудпасчера выступают лёгочное кровотечение и прогрессирующий гломерулонефрит. Статистические данные. Распространённость: 0,5 на 1 000 000. Возраст: 5–40 лет. Преобладающий пол — мужской (6:1).
Этиология неизвестна. К факторам риска относят: курение, респираторную инфекцию, контакт с летучими углеводородами.
Генетические аспекты. Выявлена ассоциация с HLA DRw2.
Патоморфология • Почки •• Эпителиальноклеточные полулуния •• Сморщивание почечных клубочков •• Интерстициальный воспалительный экссудат • Лёгкие •• Внутриальвеолярные кровоизлияния •• Макрофаги, нагруженные гемосидерином •• Фиброз межальвеолярных перегородок.
Клиническая картина • Поражение лёгких (опережает поражение почек на несколько недель или месяцев); варьирует от небольшой одышки до повторных лёгочных кровотечений с развитием дыхательной недостаточности • Быстропрогрессирующий гломерулонефрит с развитием почечной недостаточности • Артериальная гипертензия (20%) • Гриппоподобный синдром: лихорадка, миалгии, артралгии, слабость.
Лабораторные данные • ОАК •• гипохромная ЖДА • ОАМ •• микрогематурия •• протеинурия, не достигающая нефротического порога • АТ против Аг базальной мембраны клубочков почек (радиоиммунный метод или метод непрямой иммунофлюоресценции) • Перинуклеарные антинейтрофильные АТ.
Инструментальные данные • Биопсия почек — отложения иммуноглобулинов и комплемента в базальной мембране клубочков, экстракапиллярные полулуния в 50% клубочков • Рентгенография органов грудной клетки — летучие асимметричные облаковидные инфильтраты; деструкция лёгких не характерна.
Дифференциальная диагностика • Гранулематоз Вегенера протекает с деструкцией лёгочной ткани и поражением верхних дыхательных путей • Микроскопический полиангиит, помимо лёгочного и почечного синдромов, имеет и другие (множественный мононеврит, кожные изменения) • Тромбоз почечных вен и ТЭЛА (острое развитие, отсутствие иммунологической активности) • Быстропрогрессирующий гломерулонефрит, осложнённый отёком лёгких.
ЛЕЧЕНИЕ
Общая тактика предполагает сочетание плазмафереза, активной иммунодепрессивной терапии и методов коррекции почечной функции. Диета соответствует диете при почечной недостаточности.
Лекарственное лечение • ГК •• Пульс-терапия метилпреднизолоном 30 мг/кг/сут в/в капельно в течение 20–30 мин 3 дня подряд •• Преднизолон после пульс-терапии в дозе 2 мг/кг внутрь до клинического эффекта с постепенным снижением дозы: до 1,75 мг/кг в течение 1 мес, 1,5 мг/кг в течение 3 мес, далее каждую дозу назначают по 6 мес (1,25–1,0–0,75–0,5–0,25 мг/кг), далее каждую дозу назначают по 12 мес (0,125–0,0625 мг/кг). Больным старше 60 лет дозу следует снизить на 25%.
• Цитотоксические иммунодепрессанты применяют в комбинации с ГК •• Циклофосфамид 2–3 мг/кг/сут •• Азатиоприн 1–2мг/кг/сут. Примечания: дозу препарата следует уменьшить на 50% при снижении СКФ до 10 мл/мин; необходим контроль за количеством лейкоцитов и тромбоцитов крови.
Немедикаментозная терапия • Плазмаферез ежедневно или через день в сочетании с иммунодепрессивной терапии в течение 1–2 нед • Гемодиализ — при развитии почечной недостаточности.
Хирургическое лечение • Трансплантация почки в терминальной стадии ХПН. Рецидива заболевания в трансплантате не наблюдают, если перед операцией в крови не обнаруживались АТ к базальной мембране клубочков.
Прогноз неблагоприятен, если на момент постановки диагноза (до начала лечения) уже имеются признаки почечной недостаточности.
Синдром Гудпасчера: причины, симптомы, диагностика, лечение

Весь контент iLive проверяется медицинскими экспертами, чтобы обеспечить максимально возможную точность и соответствие фактам.
У нас есть строгие правила по выбору источников информации и мы ссылаемся только на авторитетные сайты, академические исследовательские институты и, по возможности, доказанные медицинские исследования. Обратите внимание, что цифры в скобках ([1], [2] и т. д.) являются интерактивными ссылками на такие исследования.
Если вы считаете, что какой-либо из наших материалов является неточным, устаревшим или иным образом сомнительным, выберите его и нажмите Ctrl + Enter.

Синдром Гудпасчера – аутоиммунный синдром, включающий альвеолярное легочное кровотечение и гломерулонефрит, вызванный циркулирующими анти-GBM-антителами. Синдром Гудпасчера чаще всего развивается у людей с наследственной предрасположенностью, которые курят сигареты, но возможными дополнительными факторами являются вдыхание углеводорода и вирусные инфекции дыхательных путей. Симптомы синдрома Гудпасчера включают одышку, кашель, усталость, кровохарканье и/или гематурию. Синдром Гудпасчера подозревается у пациентов с кровохарканьем или гематурией и подтверждается наличием анти-GВМ-антител в крови. Лечение синдрома Гудпасчера включает плазмаферез, глюкокортикоиды и иммунодепрессанты типа циклофосфамида. Прогноз благоприятен, если лечение начато до развития дыхательной или почечной недостаточности.
Синдром Гудпасчера впервые описано Гудпасчером в 1919 году. Синдром Гудпасчера представляет собой комбинацию гломерулонефрита и альвеолярного кровотечения в присутствии анти-GВМ-антител. Синдром Гудпасчера чаще всего проявляется сочетанием диффузного альвеолярного кровотечения и гломерулонефрита, но иногда вызывает изолированный гломерулонефрит (10-20 %) или поражение легких (10 %). Мужчины болеют чаще, чем женщины.

[1], [2], [3], [4], [5]
Что вызывает синдром Гудпасчера?
Причина заболевания точно не установлена. Предполагается генетическая предрасположенность к синдрому Гудпасчера, ее маркером считают наличие HLA-DRW2. Существует точка зрения о возможной роли перенесенной вирусной инфекции (вирус гепатита А и др. вирусные заболевания), производственных вредностей, лекарственных препаратов (прежде всего D-пеницилламина).
Основой патогенеза синдрома Гудпасчера является образование аутоантител к базальным мембранам капилляров клубочков почек и альвеол. Эти антитела относятся к классу IgG, они связываются с антителами базальных мембран в присутствии С3-компонента комплемента с последующим развитием иммунного воспаления почек и альвеол легких.
Анти-GВМ-антитела направлены против неколлагенового (NC-1) домена аЗ цепи коллагена IV типа, который в наиболее высокой концентрации находится в базальных мембранах почечных и легочных капилляров. Воздействие экологических факторов – курения, вирусных ОРЗ и вдыхания гидрокарбонатных взвесей (более часто) – и, менее часто, пневмонии активизирует презентацию антигенов альвеолярных капилляров циркулирующим антителам у людей с наследственной предрасположенностью (наиболее часто это носители HLA-DRwl5, – DR4 и – DRB1 аллелей). Циркулирующие анти-GВМ-антитела связываются с базальными мембранами, фиксируют комплемент и вызывают клеточный воспалительный ответ, приводящий к развитию гломерулонефрита и/или легочного капиллярита.
Вероятно, существует определенная общность ауто-антигенов базальной мембраны капилляров клубочков почек и альвеол. Аутоантиген формируется под влиянием повреждающего действия этиологического фактора. Неизвестный этиологический фактор повреждает и модифицирует структуру базальных мембран почек и легких. Экскреция образовавшихся продуктов деградации базальных мембран клубочков почек при их поражении замедляется и уменьшается, что, естественно, создает предпосылки для развития аутоиммунного поражения почек и легких До сих пор окончательно неизвестно, какой компонент базальной мембраны становится ауто-антигеном. В настоящее время предполагается, что это внутренний структурный компонент базальной мембраны клубочка почки а3-цепь коллагена 4 типа.
Сформировавшиеся иммунные комплексы откладываются вдоль базальных мембран капилляров клубочков, что приводит к развитию иммуновоспалительного процесса в почечном клубочке (гломерулонефрит) и альвеолах (альвеолит). Основными клетками, принимающими участие в развитии этого иммунного воспаления, являются Т-лимфоциты, моноциты, эндотелиоциты, полиморфноядерные лейкоциты, альвеолярные макрофаги. Взаимодействие между ними обеспечивается молекулярными медиаторами, цитокинами (факторы роста – тромбоцитарный, инсулиноподобный, b-трансформирующий; интерлейкин-1, фактор некроза опухоли и др ). Большую роль в развитии иммунного воспаления играют метаболиты арахидоновой кислоты, свободные радикалы кислорода, протеолитические ферменты, адгезивные молекулы.
В развитии альвеолита при синдроме Гудпасчера огромное значение имеет активация альвеолярных макрофагов. В активированном состоянии они выделяют около 40 цитокинов. Цитокины I группы (хемотаксины, лейкотриены, интерлейкин-8) усиливают поступление полиморфноядерных лейкоцитов в легкие. Цитокины II группы (факторы роста – тромбоцитарный, макрофагальный) способствуют перемещению в легкие фибробластов. Альвеолярные макрофаги продуцируют также активные формы кислорода, протеазы, повреждающие легочную ткань.
Синдром Гудпасчера : симптомы, лечение, клинические рекомендации
Гарвардский патологоанатом Э. Гудпасчер в 1919 г. впервые описал изменения почек и легких после перенесенного гриппа, рецидивирующего кровохарканья, и появившихся двусторонних легочных инфильтратов, сопровождающихся развитием анемии у 18-летнего юноши. Позже патология была названа синдромом Гудпасчера (СГ). СГ – редкое аутоиммунное заболевание. Частота заболеваемости СГ у взрослых составляет 1 пациент на 1 млн населения в год. В качестве этиологии приводят сочетание генетической предрасположенности и влияние факторов внешней среды. Но до конца факторы, вызывающие данный синдром, не выявлены. Заболевание имеет характерную клиническую симптоматику, включающую в себя прогрессирующий гломерулонефрит и поражение легких. Диффузный некроз альвеол и альвеолярные геморрагии сопровождаются рецидивирующим кровохарканием. В связи с редкой встречаемостью в клинической практике этого аутоиммунного заболевания врачи сталкиваются с трудностями диагностики и определением дальнейшей тактики ведения пациента. В качестве специфических лабораторных маркеров данного заболевания считают антитела к базальной мембране клубочков (аБМК). Современные методы лечения значительно улучшили прогноз заболевания. По данным различных исследований, выживаемость составляет до 90%, однако тяжелые случаи заканчиваются летальным исходом. В статье описывается клинический случай молниеносно протекающего синдрома Гудпасчера у пациентки 66 лет с летальным исходом.

1. Мухин Н.А. Синдром Гудпасчера: патогенез, диагностика, лечение. М: Фарматека, 2011. № 18. С. 8-14.
2. Ohlsson S., Herlitz H., Lundberg S., Selga D., Mölne J., Wieslander J. Segelmark M. Circulating Anti-Glomerular Basement Membrane Antibodies With Predominance of Subclass IgG4 and False-Negative Immunoassay Test Results in Anti-Glomerular Basement Membrane Disease. Am. J. Kidney Dis. 2014.Vol. 63. P. 289-293.
3. Touzot M., Poisson J., Faguer S., Ribes D., Cohen P., Geffray L., Anguel N., François H., Karras A., Cacoub P., Durrbach A., Saadoun D. Rituximab in anti-GBM disease: A retrospective study of 8 patients. J. Autoimmun. 2015. Vol .60. P. 74-79.
4. Галански М., Деттмер З., Кеберле М., Оферк Я.П., Ринге К. Лучевая диагностика. Грудная клетка. М.: МЕДпресс-информ, 2013. С. 384.
5. Нефрология. Национальное руководство. Краткое издание под ред. Н. А. Мухина. М.: ГЭОТАР-Медиа, 2016. С. 23-30.
6. Буланова М.Л., Потапов Д.В., Буланов Н.М. Атипичное течение болезни Гудпасчера: клиническое наблюдение и обзор литературы // Терапевтический архив. 2018. № 6. С.130- 136.
7. Huart A., Josse A.G., Chauveau D., Korach J.M., Heshmati F., Bauvin E., Cointault O., Kamar N., Ribes D., Pourrat J., Faguer S. French Society of Hemapheresis. Outcomes of patients with Goodpasture syndrome: A nationwide cohort-based study from the French Society of Hemapheresis. J. Autoimmun. 2016. no 73. P. 24-29.
8. West S.C., Arulkumaran N., Ind P.W., Pusey C.D. Pulmonary-renal syndrome: a life threatening but treatable condition. Postgrad. Med. J. 2013. V. 89. no 1051. P. 274-283.
9. Segelmark M., Dahlberg P., Wieslander J. Anti-GBM disease with a mild relapsing course and low levels of anti-GBM autoantibodies. Clin. Kidney J. 2012. Vol. 5. no 6. P. 549-551.
Болезнь Гудпасчера – тяжелое поражение легких и почек по типу быстропрогрессирующего нефрита. В дальнейшем был дифференцирован почечно–легочный синдром, вызываемый антителами к базальной мембране клубочков почек (анти-БМК-Ат) [1; 2]. Совместное поражение легких и почек обусловлено перекрестной реакцией анти-БМК почек и антигена БМ альвеол легких. Это позволило выделить истинный синдром Гудпасчера среди системных васкулитов и разработать рациональную схему лечения. Прогноз при этом заболевании, как правило, тяжелый, требует неотложных лечебных мероприятий.
Пик заболеваемости приходится на третье десятилетие жизни, после 60 возможна вторая волна.
Этиология до конца неясна. Считается, что спровоцировать развитие могут:
вирусы (в т.ч. вирус гриппа А2), грибковые инфекции; углеводороды, некоторые лекарства (алемтузумаб); литотрипсия; генетическая предрасположенность; различные факторы внешней среды; курение.
По результатам различных исследований установлена связь клинических исходов болезни с титром афинности и подкласса антител к БМК. Антинейтрофильные цитоплазматические антитела (АНЦА) присутствуют у 10% пациентов. Присутствие антител ассоциируется с более тяжелым течением заболевания и более высокой смертностью.
Поражения легких и почек при СГ развиваются практически одновременно. Альвеолярное кровотечение – одно из грозных осложнений. Кашель и кровохаркание являются начальными признаками кровотечения, которое может быть массивным и быстро приводить к летальному исходу. Аускультативно в легких регистрируют крепитацию, начиная с базальных отделов. По мере увеличения объема крови в альвеолах процесс распространяется выше [2]. Постепенно крепитацию заменяют влажные хрипы. Нарастающие признаки дыхательной недостаточности, сохраняющиеся даже в покое, за счет выраженной гипоксемии могут приводить к нарушению сознания.
При СГ возможно поражение легких по типу фиброзирующего альвеолита. В данной ситуации тяжесть дыхательной недостаточности обусловлена интерстициальной пневмонией и прогрессирующим интерстициальным фиброзом [3].
На рентгенограммах легких видны характерные двусторонние рыхлые инфильтраты, распространяющиеся из прикорневых отделов к периферии. Диффузное затемнение отмечается во время легочного кровотечения (интроальвеолярное кровотечение) [4].
Характерным является присутствие в мокроте макрофагов, содержащих гемосидерин. В результате легочного кровотечения развивается железодефицитная анемия. Анализ мочи характеризуется наличием протеинурии, гематурии, цилиндрурии, с выраженностью в той или иной степени, реже – азотемии. Часто заболевание сопровождает прогрессирующая почечная недостаточность на фоне олигурии и даже анурии [5]. Гистологически – в легких обнаруживаются внутриальвеолярные кровоизлияния, утолщения альвеолярных перегородок и макрофаги, содержащие гемосидерин. В почках выявляется распространенный гломерулонефрит или очаговый гломерулит [5].
Когда СГ развивается быстро, в легких патологический процесс отличается массивностью поражения. В легких выявляются преимущественно свежие изменения в виде кровоизлияний от мелких до обширных, изолированных или сливающихся фокусов и полостей распада. У таких пациентов проявления пневмосклероза и гемосидероза выражены слабо. На первый план очаговый или диффузный гемосидероз и фиброз ткани органа выступают, если легочная патология нарастает медленно [6].
Обязательным для подтверждения диагноза является обнаружение в крови антител к базальным мембранам клубочков почек. Обнаружение циркулирующих и фиксированных антител к базальным мембранам почек, перекрестно реагирующих с антигенами базальных мембран легких, является доказательством аутоиммунного генеза заболевания [7].
Прогноз, как правило, неблагоприятный. Летальный исход наступает в ближайшие 6-12 месяцев от дебюта заболевания, в результате развития легочно-сердечной или почечной недостаточности [8; 9]. В связи с редкой встречаемостью в клинической практике этого аутоиммунного заболевания каждый случай СГ представляет большой теоретический и практический интерес.
Цель исследования – продемонстрировать особенности течения молниеносной формы СГ и сложности диагностики с целью своевременности постановки диагноза и определения тактики ведения пациента с данной формой заболевания.
Материалы и методы исследования
В качестве клинического примера представлен случай наблюдения пациента с молниеносно протекающим синдромом Гудпасчера.
Больная Д., 66 лет, 05.07.2018 года поступила в стационар экстренно с жалобами на выраженную общую слабость, головные боли, отёки нижних конечностей, сухость во рту, жажду, никтурию до 8-10 раз, периодически гематурию.
Из анамнеза: гипертоническая болезнь много лет, с повышением АД выше 200 мм рт. ст. Не обследовалась, лечилась амбулаторно. Постоянно принимает гипотензивные препараты (сартаны). Дважды в год проходила профилактические осмотры с полным лабораторным контролем. Во время осмотра осенью 2017 года патологии не выявлено. В 2018 г. часто болела вирусными инфекциями. Неоднократно отмечались тонзиллиты, последний сопровождался приемом антибактериальных препаратов. В течение последнего месяца отмечает снижение толерантности к физической нагрузке, нарастание общей слабости, появление жажды, снижение диуреза. В течение последней недели нарастает никтурия.
Амбулаторно, по данным лабораторных исследований, впервые диагностируются: азотемия до 792 мкм/л, анемия лёгкой степени, гиперкалиемия.
На фоне резкого ухудшения состояния в отделении терапии экстренно переведена в ОРИТ, где на фоне инфузионной и диуретической терапии восстановлен диурез. В самочувствии отмечалась положительная динамика. В лабораторных данных наметилась тенденция к снижению уровня азотемии. По данным УЗИ органов брюшной полости, признаки нефропатии, размеры почек в пределах нормы.
По достижении стабилизации состояния пациентка переведена в отделение терапии для дообследования и лечения.
При клиническом осмотре в отделении состояние больной оценивалось как среднетяжелое. Сознание ясное. Положение активное. Телосложение правильное. Костно-мышечная система без видимой патологии. Подкожно-жировой слой выражен умеренно. Кожные покровы влажные, чистые, бледные. Видимые слизистые чистые. Язык влажный, обложен белым налетом. Периферические лимфоузлы не пальпируются. Грудная клетка обычной формы, симметричная, безболезненная при пальпации. Перкуторно над легкими ясный лёгочный звук. Дыхание ослаблено, хрипов нет, частота дыхания – 20 в минуту. Область сердца не изменена. Верхушечный толчок в V межреберье. Границы относительной сердечной тупости: правая – IV межреберье по правому краю грудины, левая – на 2 см кнаружи от левой грудино-ключичной линии в V межреберье, верхняя – III межреберье. Тоны сердца звучные, ритмичные. ЧСС-78 уд/мин., Ps 78 в мин. АД-210/90 мм рт. ст. Живот при пальпации мягкий, безболезненный. Печень не выступает из-под края реберной дуги, селезенка не пальпируется. Физиологические отправления: в норме. Периферических отёков нет.
В связи с данной симптоматикой 09.07.18 года пациентка была проконсультирована главным нефрологом города, но диагноз оставался неясен. Нефрологом было рекомендовано проведение диализа до уточнения патологии почек. Также рекомендовалось исключить урологическую, гинекологическую и онкопатологию. Через сутки наступило ухудшение самочувствия в виде нарастания дыхательной недостаточности, недостаточности кровообращения, в связи с чем больная дальнейшего лечения была переведена в ОРИТ. При проведении сеанса гемодиализа у пациентки отмечались: эпизод кровохарканья, лёгочное кровотечение с формированием двусторонней субтотальной пневмонии, гематурия, лабораторно выраженное нарушение коагуляции крови. На основании выше перечисленных данных был заподозрен синдром Гудпасчера. По жизненным показаниям назначена пульс-терапия метипредом 1000 мг в сочетании с плазмаферезом, а также взят анализ крови на АНЦА, анти-БМК.
Результаты исследований и их обсуждение
Анализ крови на RW, HBsAg, HCVAg № 713978 от 05.07.2018 г. – отрицат., кровь на ВИЧ № 70 от 10.07.2018 г. – отрицат. Анализ крови на эритропоэтин от 06.07.18 г.: 5.07 мМЕ/мл.
Анализ крови на ГЛПС № 17-1682 от 06.07.18 г. – отрицат. Антитела класса IgG к базальной мембране клубочков почек 12.07.18 г.: >200.00 Ед/мл. Анализ крови на АNА от 12.07.18 г.: отрицат. Анализ крови на АНЦА от 12.07.18 г.: 12.46 Ед/мл.
Анализ крови на парапротеины от 10.07.18 г.: моноклональной секреции не выявлено.

